Importancia de los Programas de Cribado Neonatal (PCN)
Los Programas de Cribado Neonatal (PCN), comúnmente conocidos como la prueba del talón, representan una herramienta fundamental en la salud pública. Su principal objetivo es la identificación y el tratamiento precoz de una serie de enfermedades endocrino-metabólicas que cumplen con criterios de inclusión específicos. Esta prueba debe ofrecerse a todos los recién nacidos, asegurando que las familias sean informadas previamente sobre su propósito y beneficios.

Estos programas permiten establecer un diagnóstico temprano en el recién nacido, posibilitando la implementación de un tratamiento y/o la realización de una intervención sanitaria, a menudo antes de la aparición de los síntomas de la enfermedad. Esta precocidad es crucial para reducir la morbilidad, la mortalidad y la discapacidad asociadas a estas patologías. El beneficio principal del programa de cribado neonatal es, por tanto, la prevención de discapacidades ligadas a la enfermedad. De no llevarse a cabo este cribado neonatal, y según el tipo de dolencia, se pueden dar casos que produzcan la eliminación o reducción significativa de la morbilidad, discapacidades severas y, en casos más graves, la muerte. Por lo tanto, la no realización de este cribado puede marcar la diferencia entre que un niño pueda llevar una vida normal o padezca secuelas de por vida.
El Cribado Neonatal en España: Panorama Actual y Desafíos
Cartera Común y Autonomía de las Comunidades
En España, el Ministerio de Sanidad ha establecido una cartera común básica de enfermedades endocrino-metabólicas que deben incluirse en todos los programas de cribado neonatal de las distintas comunidades autónomas. No obstante, debido a la descentralización de las responsabilidades de Salud Pública, las comunidades autónomas tienen la facultad de decidir la inclusión de más enfermedades en sus respectivos programas.
A lo largo de los últimos años, diversas comunidades autónomas han ido incorporando progresivamente más enfermedades a sus programas de cribado. Algunos ejemplos de enfermedades de inclusión reciente son el déficit de biotinidasa, la inmunodeficiencia combinada grave (SCID) y la atrofia muscular espinal (AME).
Clasificación General de Enfermedades Cribadas
Las enfermedades endocrino-metabólicas incluidas en los PCN se clasifican en grupos según su fisiopatología:
- Trastornos del metabolismo de los aminoácidos o aminoacidopatías, como la fenilcetonuria.
- Trastornos del metabolismo de los ácidos orgánicos o acidurias orgánicas, como la aciduria glutárica de tipo I.
- Trastornos del metabolismo de los ácidos grasos o defectos congénitos de la beta oxidación, como la deficiencia de acil-CoA deshidrogenasa de cadena media (MCADD).
- Otras patologías endocrino-metabólicas, como la fibrosis quística o el hipotiroidismo congénito.
Debate sobre Nuevas Inclusiones
La inclusión de patologías como la inmunodeficiencia combinada grave y la atrofia muscular espinal ha generado un importante debate. El cribado de la SCID se inició en Cataluña en enero de 2017, convirtiéndose en la primera región europea en incluir esta patología de manera oficial y universal. Varias comunidades autónomas se han sumado desde entonces. La aprobación por parte del Ministerio de Sanidad de nuevos tratamientos para la AME ha evidenciado la necesidad de cribar esta patología para ofrecer un tratamiento rápido y presintomático. Consecuentemente, diversas comunidades ya han aprobado su cribado neonatal, mientras otras realizan estudios piloto para demostrar su coste-efectividad.
Desafío de la Falta de Consenso y Derechos Constitucionales
En España, aún no existe un consenso uniforme entre las Comunidades Autónomas sobre las enfermedades a incluir en el Programa de Detección Precoz de Metabolopatías. Esta disparidad implica que la supervivencia y la calidad de vida de los recién nacidos españoles pueden depender de su lugar de nacimiento. Ejemplos como la Acidemia Metilmalónica, entre otras metabolopatías, ya se detectan en muchas Comunidades Autónomas, confirmando sus beneficios terapéuticos. Para estas, solo sería necesario replicar procedimientos ya implementados en otras regiones, sin necesidad de estudios de viabilidad o costes económicos adicionales, dada la tecnología existente en los laboratorios.
Asociaciones como ACIMET y la FEEMH han denunciado casos de diagnóstico tardío debido a la no inclusión de ciertas enfermedades en el cribado neonatal. Recuerdan que el artículo 43 de la Constitución española reconoce "el derecho a la protección de la salud" y que "compete a los poderes públicos organizar y tutelar la salud pública a través de medidas preventivas y de las prestaciones y servicios necesarios". Asimismo, el artículo 15 afirma que "Todos tienen derecho a la vida y a la integridad física y moral". Por ello, estas entidades solicitan a las administraciones trabajar urgentemente en la inclusión de todas aquellas enfermedades metabólicas cuyo cribado ya se realiza en otras comunidades y para las que existe evidencia suficiente de coste-efectividad y pertinencia.
La Homocistinuria: Una Enfermedad Metabólica Hereditaria
Definición y Causas Principales
La homocistinuria (HCU) es una enfermedad congénita hereditaria poco frecuente que, si no se trata, puede provocar complicaciones graves. Se debe a errores en el gen responsable de crear una enzima clave llamada cistationina beta sintasa (CBS). Los pacientes con homocistinuria presentan una deficiencia de esta enzima y, como resultado, tienen niveles altos de una sustancia llamada homocisteína en sus cuerpos.
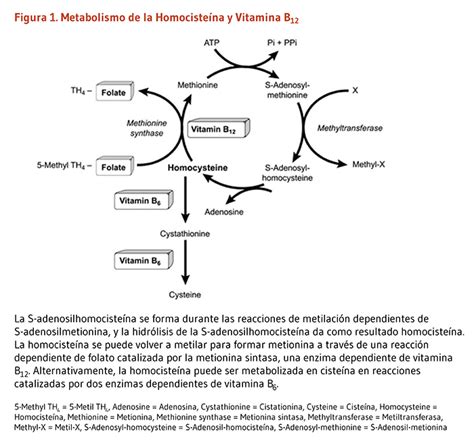
Esta enfermedad afecta la capacidad de un bebé para metabolizar el aminoácido metionina, un componente proteico esencial. Esto conduce a un aumento anormal en los niveles de metionina y homocisteína. La homocistinuria es la causa más frecuente de hiperhomocisteinemia, una elevada concentración de homocisteína en plasma y orina, resultado de la acumulación tóxica de este aminoácido en los tejidos.
Tipos de Homocistinuria por Deficiencia de CBS
Existen diferentes formas de homocistinuria, todas caracterizadas por concentraciones elevadas de metionina u homocisteína. En la forma más común, la "homocistinuria por deficiencia de CBS", el cuerpo no produce correctamente la enzima CBS, que utiliza vitamina B-6 para convertir la homocisteína en otros aminoácidos necesarios. Dentro de esta forma, se distinguen dos tipos:
- Homocistinuria que responde a la vitamina B-6: La enzima CBS está dañada, pero puede utilizar la vitamina B-6 para funcionar.
- Homocistinuria que no responde a la vitamina B-6: La enzima CBS está dañada y no puede utilizar la vitamina B-6.
En ambas formas, el organismo tiene dificultades para eliminar la homocisteína, lo que lleva a su acumulación y a la de metionina.
Fisiopatología y Consecuencias Clínicas
Aunque los pacientes con homocistinuria parecen normales al nacer, durante un período de meses y años desarrollan problemas graves. Los niveles elevados de homocisteína y metionina pueden causar coágulos de sangre y dañar el cerebro y los ojos. Las complicaciones incluyen problemas óseos y oculares, retraso del desarrollo mental, discapacidades intelectuales y anomalías vasculares. La acumulación de homocisteína daña el endotelio, la capa de células especializadas que reviste el interior de los vasos sanguíneos y el corazón, lo que se asocia a una temprana aterogénesis y a un mayor riesgo de enfermedad coronaria, vascular cerebral y periférica. Los episodios tromboembólicos son la principal causa de mortalidad en estos pacientes.
Día Mundial de Concienciación y Esfuerzos de Asociaciones
Cada 18 de mayo se celebra el Día Mundial de Concienciación sobre la Homocistinuria, destacando su naturaleza como enfermedad metabólica hereditaria poco frecuente. La Asociación Española para el Estudio de los Errores Innatos del Metabolismo (AECOM), fundada en 1999, trabaja activamente en aglutinar a profesionales dedicados al diagnóstico, tratamiento e investigación en los errores congénitos del metabolismo. Recientemente, la revista 'Nutrients' publicó un trabajo sobre la hiperhomocisteinemia en adultos, promovido por AECOM con la colaboración de Recordati Rare Diseases, que contó con la participación de expertos nacionales de diversos centros de referencia.
Como indican estos expertos, la hiperhomocisteinemia es una condición tratable que requiere un enfoque multidisciplinario para su detección, manejo y prevención de complicaciones, con el objetivo de mejorar la calidad de vida de los pacientes adultos afectados. Advierten que el diagnóstico de los trastornos que afectan el metabolismo de la homocisteína afronta demoras debido al conocimiento insuficiente de su presentación clínica y a unas características bioquímicas únicas.
El Cribado Neonatal de Homocistinuria: Diagnóstico y Tratamiento Precoz
Métodos Actuales de Detección
El cribado neonatal de homocistinuria es de vital importancia para su diagnóstico temprano, ya que la enfermedad puede tener graves consecuencias si no se detecta y aborda precozmente. El cribado se realiza mediante la determinación de metionina en una muestra de sangre obtenida del talón del recién nacido durante las primeras 24-72 horas de vida. Estas gotas de sangre se envían para su análisis a un laboratorio especializado en cada Comunidad Autónoma. Las familias, como norma general, reciben los resultados en su domicilio. Si se detecta alguna enfermedad, se les informa para poder establecer un tratamiento.
Nuevas Perspectivas en el Cribado: Medición Directa de Homocisteína
6ª. Jornada de Tamizaje Neonatal
Las pruebas actuales solo miden los niveles de metionina, los cuales pueden permanecer bajos durante la evaluación de recién nacidos, lo que puede llevar a diagnósticos tardíos. Científicos de los Centros para el Control y la Prevención de Enfermedades (CDC, Atlanta, GA, EUA) han desarrollado una nueva prueba que podría mejorar notablemente la calidad de vida de los bebés con HCU. En los recién nacidos con HCU, los niveles de homocisteína presentan un aumento temprano y significativo, generalmente antes que los niveles de metionina. Esto convierte a la homocisteína en un marcador ideal para detectar la enfermedad durante los primeros días de vida. El equipo de investigación evaluó el rendimiento de esta prueba en muestras residuales de detección de recién nacidos, demostrando su eficacia al distinguir efectivamente entre muestras sanas y HCU-positivas. Este método es el único de primer nivel de análisis de inyección de flujo y espectrometría de masas en tándem que cuantifica directamente la homocisteína total a partir de gotas de sangre seca.
Tratamiento y Pronóstico
Detectar la homocistinuria en esta etapa temprana permite iniciar el tratamiento de forma precoz, lo que ayuda a reducir los niveles de homocisteína en sangre y prevenir complicaciones graves como discapacidad intelectual y episodios tromboembólicos. El tratamiento con una dieta especial y fármacos puede prevenir el desarrollo de estas complicaciones, pero debe comenzar en los primeros años de vida para ser verdaderamente efectivo. El objetivo del tratamiento es prevenir los problemas de salud que causa esta enfermedad. Los tratamientos pueden incluir:
- Dieta baja en proteínas (específicamente metionina).
- Preparados y alimentos especiales.
- Vitamina B-6, betaína, ácido fólico u otros suplementos vitamínicos.
- Medicamentos para ayudar a eliminar el exceso de homocisteína.
Los niños que reciben un tratamiento temprano y continuo para la homocistinuria pueden gozar de un crecimiento y desarrollo saludables. El tratamiento es muy importante para reducir la posibilidad de que se produzcan coágulos de sangre y problemas oculares y cardíacos.
Evidencia Científica sobre el Cribado de Homocistinuria
Revisión Cochrane y Necesidad de Estudios Futuros
Una revisión Cochrane actualizada, que evaluó la evidencia hasta el 8 de junio de 2015, revisó si el cribado de la población de recién nacidos para el diagnóstico de homocistinuria por deficiencia de cistationina beta sintasa conduce a una mejoría clínica en comparación con el diagnóstico clínico posterior, y determinó los efectos psicológicos en los padres o cuidadores. En esta revisión, no se encontró ningún ensayo controlado aleatorizado ni ensayos clínicos controlados que evaluaran el uso de cualquier prueba de detección precoz neonatal para diagnosticar a los recién nacidos con homocistinuria antes de que la enfermedad sea clínicamente evidente. Por lo tanto, no es posible establecer ninguna conclusión basada en estudios controlados.
Sin embargo, se conocen algunos estudios no controlados y series de casos que han indicado que los métodos de detección precoz de la homocistinuria en el recién nacido y su tratamiento precoz fueron efectivos. Estos estudios apoyan la eficacia del cribado. Se necesitan estudios de investigación futuros a largo plazo para aportar evidencia sólida a favor o en contra de los métodos de detección precoz.
tags: #cribado #neonatal #homocistinuria #regla #nemotecnica