El cribado neonatal es una herramienta fundamental de salud pública que permite la detección temprana de enfermedades congénitas y metabólicas en recién nacidos. Este proceso ha evolucionado significativamente en España, beneficiando a miles de niños y sus familias. En este contexto, la figura de Felícitas Díaz-Flores Estévez destaca por su profunda experiencia y su rol como asesora experta en el desarrollo y la calidad de estos programas.
Díaz-Flores Estévez es una reconocida experta en cribado neonatal, desempeñándose también como asesora de la Ponencia de cribado poblacional de la Dirección General de Salud Pública del Ministerio de Sanidad en Madrid. Su experiencia abarca la evaluación de la calidad del cribado neonatal, lo que subraya su compromiso con la excelencia en este campo vital.
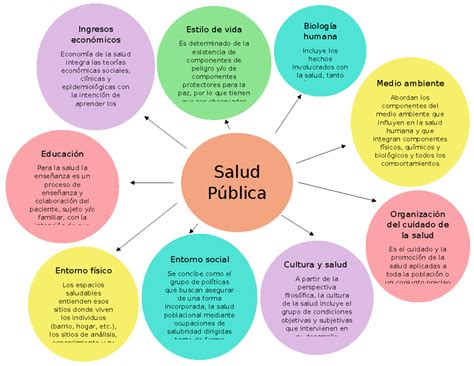
Evolución Histórica del Cribado Neonatal en España
Existe un amplio consenso sobre los beneficios en salud que ha aportado el cribado neonatal en España. Su historia se remonta a 1968, cuando el Profesor Mayor Zaragoza inició en Granada su proyecto de investigación para la detección precoz de la fenilcetonuria y otras aminoacidopatías. Estos Programas de Cribado Neonatal (PCN) se fueron desarrollando hasta los años 80 en torno al llamado "Plan Nacional de Prevención de la Subnormalidad", cubriendo aproximadamente el 30% de los recién nacidos españoles.
A partir de 1982, con el inicio de la gestión de la sanidad transferida a las Comunidades Autónomas (CCAA), los PCN se expandieron. Este periodo sentó las bases para que se convirtieran en una actividad organizada y multidisciplinar, integrada y coordinada desde el Sistema de Salud. Sin embargo, la descentralización de las responsabilidades de Salud Pública en las CCAA afectó el desarrollo de los PCN, creando diferencias entre ellas al atomizarse las decisiones sobre la ampliación de las enfermedades a cribar.
Hacia la Armonización y Estandarización de los Programas
Inicialmente, la disponibilidad de métodos de detección y tratamiento eficaces era, muchas veces, la justificación exclusiva para la inclusión de nuevas enfermedades en un PCN. Esta situación generó una enorme desigualdad en el acceso al cribado neonatal. No obstante, ha cambiado con la introducción de legislación apropiada que garantiza el correcto desarrollo de los PCN dentro del Sistema Nacional de Salud. Los foros coordinados por el Ministerio de Sanidad, con la participación de los responsables de Salud Pública de las CCAA y las sociedades científicas, han sido fundamentales en este proceso.
En los años 2005 y 2006, las Sociedades Científicas SEQC (Sociedad Española de Química Clínica) y AECNE (Asociación Española de Cribado Neonatal), con la coordinación del Área de Promoción de la Salud de la Dirección General de Salud Pública, recopilaron información y elaboraron un informe sobre los PCN en España para el Consejo Interterritorial del Sistema Nacional de Salud (CISNS). En julio de 2013, este Consejo aprobó siete enfermedades que debían formar parte del panel de detección de los PCN territoriales, un hito clave hacia la armonización de estos programas. Posteriormente, se establecieron los objetivos y requisitos de calidad del Programa de Cribado Neonatal de enfermedades endocrino-metabólicas del Sistema Nacional de Salud a nivel nacional, lo que llevó a la ampliación del cribado neonatal en toda España, incluyendo al menos ocho enfermedades.
Criterios de Inclusión y Avances Tecnológicos
Los PCN son reconocidos en los diferentes sistemas sanitarios como programas esenciales de prevención en Salud Pública y están integrados en la actividad asistencial de las unidades pediátricas. Los PCN no deben identificarse solo con un procedimiento técnico o de laboratorio, sino como una actividad coordinada del sistema sanitario que asegure su eficacia y eficiencia.
Para la inclusión de un trastorno en el cribado masivo, se han definido criterios clave:
- La gravedad de la enfermedad, que cursa con morbilidad (como retraso mental) o mortalidad si no se diagnostica en el período neonatal.
- La existencia de un tratamiento eficaz.
- Una frecuencia relativamente elevada de la enfermedad (al menos, 1 de cada 10.000-15.000 recién nacidos).
- La disponibilidad de un método analítico de cribado rápido, fiable y de bajo coste (criterios establecidos por Wilson & Jungner en 1968).
Estos criterios buscan garantizar el objetivo principal de los programas: "el máximo beneficio con el mínimo de costes". Sin embargo, con el tiempo, estos criterios han evolucionado, renovándose gracias a la nueva tecnología de Espectrometría de Masas en Tándem (MS/MS), e incluso en un contexto de cribado genético. La incorporación de esta tecnología permite la determinación de múltiples enfermedades utilizando la misma técnica analítica y una cantidad de muestra muy reducida.
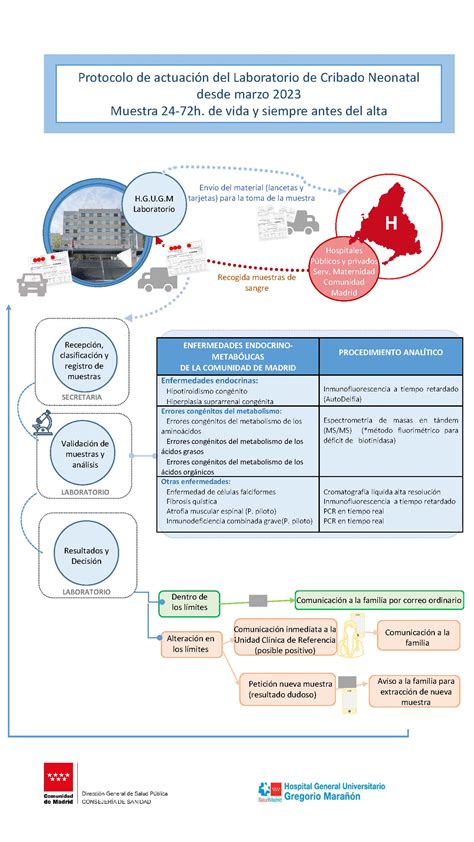
Las muestras con valores alterados en los marcadores seleccionados para su cribado requieren confirmación con una segunda muestra. Si los resultados se confirman, se considera positivo para el cribado y se deriva a la unidad clínica de referencia.
Garantía de Calidad en los Laboratorios de Cribado Neonatal
La calidad es un pilar fundamental en el cribado neonatal. Los laboratorios de cribado neonatal deben adherirse a estrictos estándares de calidad. Entre los requisitos para la competencia de los proveedores de ensayos de aptitud y los métodos estadísticos para su uso, se encuentran normas como la ISO 17043:2010 y la ISO 13528:2015. Además, el protocolo armonizado internacional para las pruebas de aptitud de laboratorios de química analítica (IUPAC 2006) y la definición de las especificaciones de rendimiento analítico son cruciales para asegurar la fiabilidad de los resultados.
Espectrómetro de Masas Triple Cuadrupolo
El Cribado Neonatal Ampliado en Canarias: Un Caso Ilustrativo
El Cribado Neonatal Ampliado (CNA) en Canarias es un ejemplo de la implementación y los desafíos de estos programas. Comenzó en el año 2015, siguiendo las directrices nacionales de 2013. Inicialmente, incluía la fenilcetonuria (PKU), hipotiroidismo congénito (HtC), fibrosis quística (FQ), aciduria glutárica tipo 1 (AG-1), déficit de acil-CoA de cadena media (MCAD), déficit de 3-hidroxiacil-CoA de cadena larga (LCHAD) y anemia de células falciformes. En septiembre de 2021, se añadió el déficit de biotinidasa, sumando un total de ocho enfermedades.
Implementación del CNA en Canarias
El Programa de Diagnóstico Precoz de Metabolopatías Congénitas en el Recién Nacido de la Comunidad Autónoma de Canarias dependió del Departamento de Pediatría de la Universidad de La Laguna desde 1981 hasta el primer trimestre de 2021. Tras la finalización del convenio, la Unidad de Cribado Neonatal de Canarias pasó a depender del Servicio Canario de Salud, adjudicándose al Hospital Universitario de Canarias después de la presentación de proyectos por parte de tres hospitales. Este cambio marcó una nueva etapa, con posibilidades de ampliación, mejoras en recursos humanos y materiales, y centralización de las muestras.
En octubre de 2021, la deficiencia de biotinidasa se incorporó al cribado mediante la determinación de su actividad por fluorimetría en la misma muestra de sangre seca en papel utilizada para las otras enfermedades. El cribado de la mayoría de estas enfermedades se realiza mediante espectrometría de masas en tándem (MS/MS).
Desafíos y Mejoras en el Diagnóstico
La condición de región ultraperiférica de Canarias ha representado una gran debilidad en el diagnóstico y seguimiento de los pacientes con metabolopatías. Hasta hace poco, las pruebas de primera línea como el perfil de aminoácidos (AA), acilcarnitinas (AC) y ácidos orgánicos (AO) no estaban disponibles en los hospitales canarios, debiendo enviarse a laboratorios nacionales. Esto implicaba tiempos de envío de muestras de 2-3 días y tiempos de respuesta de 1-3 semanas, lo que es especialmente crítico en urgencias metabólicas, donde la espera puede prolongarse hasta 5-7 días. Esta situación resultaba en un manejo casi a ciegas de enfermedades complejas y graves, con una clara desventaja en comparación con el resto del territorio nacional, llevando a una mayor morbimortalidad.
Para muchos pacientes, especialmente en los dos primeros años de vida, se requieren controles mensuales, e incluso semanales o diarios. Los largos tiempos de espera entre el envío de muestras, el análisis y la recepción de resultados impedían un control y seguimiento adecuados. A menudo, esto obligaba al traslado de pacientes a hospitales de referencia en la península (Madrid, Barcelona), con el consiguiente sobrecoste para la administración y las familias.
Actualmente, el laboratorio central del Complejo Hospitalario Universitario de Canarias trabaja para incorporar las determinaciones analíticas necesarias para la confirmación de casos positivos de cribado y el seguimiento de casos confirmados. Este avance representa un gran paso para los enfermos y especialistas en metabolopatías.
Enfermedades Específicas del Cribado: La Fenilcetonuria (PKU)
La Fenilcetonuria (PKU), descrita por primera vez en 1934 como Oligofrenia Fenilpirúvica, es una enfermedad mendeliana de herencia autosómica recesiva. Se caracteriza por un aumento de la fenilalanina (Phe) en sangre debido al déficit funcional de la enzima fenilalanina hidroxilasa (PAH), esencial para la conversión de Phe a tirosina (Tyr). Esta alteración provoca una disminución de aminoácidos largos neutros (Trp, Tyr, Hys, Met, Thr) y aminoácidos de cadena ramificada (Leu, Ile, Val), afectando tanto la absorción intestinal como los transportadores cerebrales.
La incidencia de la fenilcetonuria varía regionalmente, con una tasa en Canarias de 1:10.222 habitantes en 2016. La clínica de la PKU es multifactorial, manifestándose no solo por la elevación de la Phe sino también por fallo de medro, retraso mental, microcefalia, alteraciones del metabolismo óseo y alteraciones neuropsicológicas. Estas últimas pueden aparecer incluso con un control metabólico óptimo e incluyen trastorno por déficit de atención con hiperactividad, alucinaciones, convulsiones y alteraciones del comportamiento.
Detección de la PKU mediante Cribado Neonatal
El diagnóstico de la PKU se realiza mediante cribado neonatal. Para el cribado de la PKU, se cuantifican los niveles de fenilalanina y el cociente fenilalanina/tirosina a partir del perfil de aminoácidos. Valores de Phe superiores a 97 µmol/L y un cociente Phe/Tyr superior a 1,7 sugieren la enfermedad. Es crucial que la toma de muestra se realice después de la primera toma de alimento del recién nacido. Es importante considerar los posibles falsos positivos si el recién nacido ha recibido nutrición parenteral antes de la toma de muestra; en estos casos, se debe registrar esta información y remitir una segunda muestra 72 horas después de la interrupción de la nutrición parenteral.
Las pruebas de confirmación incluyen la determinación de aminoácidos en sangre y orina, y el estudio de pterinas en orina para descartar alteraciones en la síntesis y regeneración del cofactor. Posteriormente, se realiza el estudio de mutaciones del gen PAH para un adecuado consejo genético, utilizando bases de datos como BioPKUdb.org. En Canarias, la mutación p.R408W es la más prevalente.
El tratamiento de la fenilcetonuria se basa en un control dietético estricto de la ingesta de fenilalanina. Gracias a productos ricos en aminoácidos y exentos de fenilalanina, los pacientes pueden mantener la ingesta de nutrientes y controlar los niveles de fenilalanina en sangre.
Otras Enfermedades Detectadas por Espectrometría de Masas en Tándem (MS/MS)
Mediante MS/MS también se realiza el cribado de otras enfermedades metabólicas. Para el cribado de la MCAD (Déficit de acil-CoA deshidrogenasa de cadena media), LCHAD (Déficit de 3-hidroxiacil-CoA deshidrogenasa de cadena larga) y AG-1 (Aciduria glutárica tipo 1), se analiza el perfil de acilcarnitinas.
- Para la MCAD, se evalúan marcadores como C8, C6, C10, C10:1, C8/C10 y C8/C29.
- Para la LCHAD, los marcadores incluyen C16-OH, C18-OH, C18:1-OH, C18:2-OH, C16:1-OH, C14-OH, C14:1-OH, C16-OH/C16 y C18-OH/C18.
- Para la AG-1, se analizan C5DC y cocientes como C5DC/C8, C5DC/C16, C5DC/C0, C5DC/C5OH y C5DC/C3DC.