En neuroanatomía, la amígdala cerebelosa -denominada también tonsila cerebelosa- es la porción más inferomedial de cada hemisferio del cerebelo. Se aloja en la cara inferior del lóbulo posterior, justo por encima del foramen magno. Funcionalmente, forma parte del lóbulo floculonodular y del paleocerebelo vestibular, integrando aferencias del núcleo vestibular, la médula espinal y la oliva inferior para regular el tono postural, los reflejos vestibulooculares y la coordinación fina del tronco.
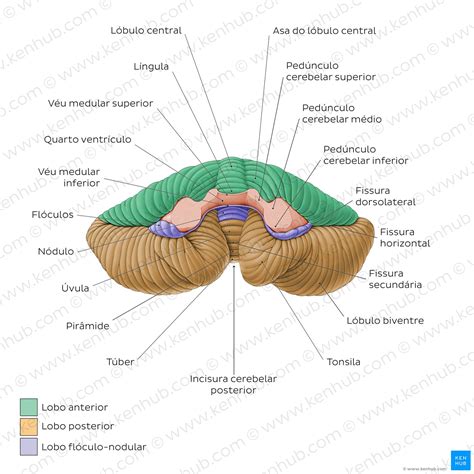
Desarrollo Embrionario de las Amígdalas Cerebelosas
El cerebelo se origina a partir de las placodas rómbicas de la pared del rombencéfalo dorsal. Durante la 4.ª-5.ª semana de gestación, los labios rómbicos proliferan y forman el istmo cerebeloso. Posteriormente, los hemisferios laterales crecen y se pliegan alrededor del vermis: la fisura posterolateral delimita los lóbulos flóculo-nodular y posterior. Las amígdalas cerebelosas se constituyen entre la 8.ª y la 12.ª semana como engrosamientos inferiores de dichos hemisferios.
La amígdala cerebelosa participa en la regulación postural adaptativa mediante un circuito triádico: estímulo vestibular → amígdala/úvula → núcleo fastigio. Al recibir aceleraciones lineales o cambios de inclinación, modula la excitabilidad de motoneuronas antigravitatorias espinales (por medio de vías vestíbulo-espinales) y ajusta los movimientos oculares (reflejo vestibulo-ocular).
¿Qué es la Ectopia Amigdalar Cerebelosa o Malformación de Chiari?
La ectopia de las amígdalas cerebelosas se define por el descenso de estas estructuras por debajo del foramen magno. Históricamente, se consideraba un descenso de 5 mm o más como criterio diagnóstico. Sin embargo, actualmente se considera que incluso descensos mínimos (2-3 mm) o incluso la ausencia de descenso, pero con un claro compromiso del espacio y patologías secundarias como la siringomielia, pueden ser indicativos de una malformación de Chiari, lo que se conoce como Chiari tipo 0.
La malformación de Arnold Chiari es una enfermedad neurológica rara y crónica en la cual el cerebelo se desplaza hacia el canal espinal. Esto sucede cuando la sección del cráneo que alberga el cerebelo (fosa posterior) es más pequeña de lo habitual y, como consecuencia, ejerce presión sobre el cerebro y lo empuja, haciendo que la parte inferior del cerebelo (amígdalas cerebelosas) se desplacen hacia el conducto medular.
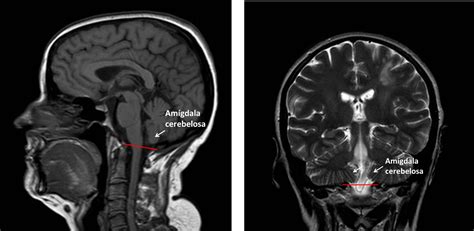
Incidencia y Origen
La incidencia de la malformación de Arnold-Chiari de Grado I se estima entre el 0,6 y el 0,9%, con una predominancia de 2:3 en hombres sobre mujeres, aunque otros estudios sugieren que afecta aproximadamente al 0,5% de la población, mayormente mujeres. El diagnóstico se realiza comúnmente entre los 30 y 40 años de edad. Es una malformación con la que se puede nacer o que puede desarrollarse con el tiempo.
Aunque se sugiere un origen genético, incluso ligado al cromosoma X, un porcentaje de casos se ha producido tras un traumatismo severo, a menudo un latigazo cervical. En muchas ocasiones, la ectopia de las amígdalas cerebelosas se diagnostica de forma casual después de un traumatismo craneoencefálico, accidente, infección o durante el embarazo, sin que el paciente haya presentado síntomas previos.
Clasificación de la Malformación de Chiari
Se distinguen varios grados de malformación de Arnold-Chiari en función de la extensión del descenso amigdalar y sus consecuencias morfológicas:
Malformación de Chiari Tipo 0
En este tipo, los pacientes tienen siringomielia con muy poco o ningún descenso amigdalar. Se cree que ocurre debido a una alteración de la hidrodinámica del líquido cefalorraquídeo (LCR) a nivel del foramen magno, probablemente por un menor tamaño de la fosa posterior. Fue descrita en 1998 por Iskandar y colaboradores, quienes documentaron la curación de casos de siringomielia sin herniación tras la descompresión de la fosa posterior.
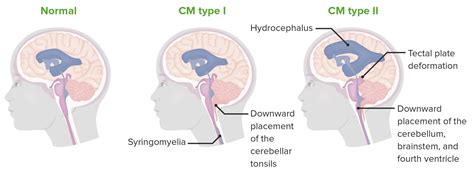
Malformación de Chiari Tipo I
Es el tipo más frecuente y consiste en el desplazamiento de las amígdalas cerebelosas, habitualmente más de 3-5 mm, en el canal cervical, sin otras malformaciones asociadas en la médula espinal. Sin embargo, un descenso menor no es excluyente y debe ser valorado. Suele dar síntomas en la segunda o tercera década de la vida, es poco frecuente en mayores de 60 años y ocurre más en mujeres. Entre el 50-70% de los afectados presentan siringomielia de localización cervical y/o dorsal, y la hidrocefalia se da en el 7-10% de los casos.
Malformación de Chiari Tipo 1.5
De reciente descubrimiento, se describe como una combinación del descenso de las amígdalas cerebelosas junto al óbex del tronco del encéfalo a través del agujero magno.
Malformación de Chiari Tipo II
En este caso, hay un defecto variable en la formación del tronco cerebral, a menudo asociada a hidrocefalia debido al bloqueo de los orificios de salida del IV ventrículo o por estrechez del acueducto. Su forma más extrema consiste en la herniación de estructuras de la porción más baja del cerebelo (amígdalas cerebelosas) y del tronco cerebral a través del foramen magno, engrosando y comprimiendo el canal espinal. Puede ir asociada a espina bífida.
Malformación de Chiari Tipo III
Es la malformación de Chiari más rara y grave, ya que además de las anomalías estructurales del tipo II, ocurre con encefalocele occipital y se asocia a graves defectos neurológicos.
Malformación de Chiari Tipo IV
El tipo IV involucra un desarrollo incompleto de las estructuras del cerebelo (hipoplasia cerebelosa) y puede incluir amígdalas muy bajas en el canal espinal, estructuras cerebelosas ausentes, y partes del cráneo y la médula espinal al descubierto.
Etiología y Teorías Asociadas
Etiología Multifactorial y Causas Adquiridas
La ectopia amigdalar puede ser de etiología multifactorial, incluyendo una fosa posterior pequeña, hipertensión venosa o disrrafismo óseo. También puede ser secundaria a otras condiciones como hematoma subdural posterior, infarto cerebeloso, tumor o hidrocefalia.
La Enfermedad del Filum y la Teoría de la Tracción Caudal
El Dr. Miguel B. Royo Salvador ha propuesto una teoría que vincula la ectopia de las amígdalas cerebelosas con la Enfermedad del Filum. Esta teoría sugiere que la tracción caudal de todo el sistema nervioso, producida por un Filum terminale excesivamente tenso, causa el mecanismo de descenso de las amígdalas cerebelosas.
Se ha observado que tras un accidente, caída o trauma de la columna vertebral -con una médula espinal que ya padece congénitamente la Enfermedad del Filum-, la tracción medular puede aumentar. En estas circunstancias de mayor tracción medular caudal, pueden observarse empeoramientos del descenso de las amígdalas cerebelosas o de la compresión en el foramen magnum, a través de los síntomas.
Malformación de Arnold Chiari (qué es , sus síntomas y tipos )
Sintomatología de la Malformación de Chiari
Los síntomas en los afectados de Arnold Chiari son muy variables de una persona a otra, dependiendo del tipo de Chiari y las patologías asociadas. No siempre se basan en la gravedad del descenso de las amígdalas cerebelosas o en la presencia de otras afecciones secundarias. Algunas personas con descensos mínimos pueden presentar numerosos síntomas, mientras que otras con descensos significativos pueden tener pocos o ninguno.
Los síntomas más comunes se relacionan con el aumento de la presión intracraneal, la afectación de los pares craneales, la compresión del tronco cerebral, la mielopatía, la afectación cerebelosa, el dolor y la siringohidromielia asociada. Entre ellos se incluyen:
- Cefalea occipital (que empeora con la maniobra de Valsalva) y dolor de cuello.
- Ataxia troncal, desequilibrio general y torpeza.
- Nistagmo (especialmente down-beat), disminución de la visión y/o visión borrosa, diplopía.
- Disestesia cervical, parestesias y/o disestesias.
- Insomnio o hipersomnia, fatiga.
- Vértigos.
- Apnea del sueño y ronquidos.
- Presión y/o zumbido en oídos, intolerancia a los sonidos.
- Dificultad de leer texto, pérdida de memoria, problemas para dar opinión en profundidad.
- Intolerancia a la luz brillante.
- Espasmos, temblores de manos.
- Náuseas.
- Problemas menstruales.
- Dificultad de caminar en superficies desiguales.
- Ansiedad y depresión.
- Dolor de presión detrás de los ojos.
- Puntos en la visión.
- Dolor generalizado.
- Sensaciones ardientes como eléctricas.
- Disfagia (dificultad para tragar), disfonía.
- Alteraciones esfinterianas.
- Déficits motores y sensitivos.
En los casos más severos, cuando se ven involucrados los pares craneales y otras patologías asociadas como la siringomielia, pueden darse atrofias linguales y del velo del paladar, parálisis de cuerdas vocales y hemiplejia, lo que puede llevar a discapacidades severas y dependencia.
Diagnóstico de la Malformación de Chiari
El diagnóstico de Chiari debería ser sencillo, ya que basta con la realización de una resonancia magnética (RM) de cráneo para detectarlo; sin embargo, en muchas ocasiones es pasado por alto debido al desconocimiento que aún existe en la población médica. Ante la presencia de alguno de los síntomas mencionados, siendo el más habitual el dolor de cabeza persistente, los pacientes deben ser estudiados de forma exhaustiva mediante un examen neurológico que incluya pruebas de imagen.
Métodos Diagnósticos
- Resonancia Magnética (RM): Es el método preferido y necesario de investigación.
- La RM craneal es la primera instancia. En ella debe observarse si existe una malformación de la unión cráneo-cervical y tomar mediciones precisas en milímetros en las imágenes de corte sagital para ver la distancia entre el foramen magno y la parte más inferior de la amígdala que descendió por debajo de él.
- Si se observan anomalías respecto al foramen magno, la cisterna magna, la fosa posterior y/o un descenso de las amígdalas cerebelosas por debajo del agujero magno, se debe pensar en una malformación de Chiari y completar el estudio con RM cervical y dorsal para descartar o confirmar siringomielia asociada.
- La RM también permite estudiar el flujo del LCR en el espacio subaracnoideo espinal y a través de los sirinx. Es vital para el seguimiento postoperatorio, para medir el tamaño de los sirinx y detectar posibles complicaciones.
- Potenciales Evocados: Somatosensoriales, auditivos y visuales, que proporcionarán información más precisa del estado neurológico y de si existen daños.
- Estudios de Radiografía: Pueden mostrar un ensanchamiento del canal espinal a nivel cervical, así como la malformación de la unión cráneo-cervical. El electromiograma y el TAC (Scanner) proporcionan un valor pequeño de información hoy día.
Patologías Asociadas a la Malformación de Chiari
Siringomielia / Siringohidromielia
La siringomielia es una enfermedad caracterizada por la presencia de cavidades quísticas dentro del cordón espinal. Es una enfermedad progresiva de forma crónica, con un curso clínico imprevisible. Se prefiere el término siringohidromielia, que incluye tanto la hidromielia (canal central dilatado) como la siringomielia (cavitación de la médula que se extiende lateralmente o independiente de la cavidad central).
- Incidencia: Su incidencia es de 8.4 nuevos casos/año/100.000 personas.
- Asociación: La siringohidromielia se asocia con la malformación de Chiari tipo I entre el 30-70% de los casos. Aproximadamente el 84% de los casos de siringomielia están asociados con malformaciones de la unión cráneo-cervical, como Chiari I, Chiari II e impresión basilar. El 1% de los casos se asocia con hidrocefalia.
- Etiología: En los casos asociados con malformaciones de la unión cráneo-cervicales, el LCR no puede fluir libremente del IV ventrículo hacia el espacio subaracnoideo. La causa más común es el descenso de las amígdalas cerebelosas por debajo del foramen magno en el Chiari.
- Tipos de Cavidades: De acuerdo a los resultados de autopsias, se distinguen tres tipos de cavidades siringomiélicas:
- Dilataciones del canal central que comunican directamente con el cuarto ventrículo.
- Dilataciones del canal central sin comunicación con el cuarto ventrículo, con un segmento libre de sirinx.
- Sirinx originados en el parénquima del cordón espinal y no comunicados con el canal central.
- Manifestaciones Clínicas: Consisten en un síndrome del cordón central, caracterizado por la pérdida de sensación en una forma segmentada que afecta el cuello, hombros y brazos. La mayoría presenta una pérdida de percepción de dolor y sensaciones de temperatura con alteración del tacto, junto con una bajada de las señales motoras neuronales (parálisis flácida que progresará a amiotrofia). No es raro que los pacientes presenten también escoliosis torácica.
- Evolución: La duración de los síntomas puede variar de 4 meses a 38 años. La edad media de diagnóstico es de 32 años. La deterioración neurológica es progresiva hasta que se realiza la cirugía. La primera señal que aparece, normalmente, es la parestesia en los miembros superiores.
- Siringobulbia: Cuando la cavidad siringomiélica afecta al bulbo raquídeo, la patología se conoce como siringobulbia. La pérdida sensorial afecta a la cara y la atrofia daña la lengua, con deterioro progresivo al tragar (disfagia), ronquidos y apneas durante el sueño.
Hidrocefalia
La hidrocefalia consiste en la acumulación excesiva de líquido cerebroespinal (LCR), resultando en la dilatación anormal de los ventrículos cerebrales y una presión potencialmente perjudicial en los tejidos del cerebro. En la malformación de Chiari II, la hidrocefalia es prácticamente constante, mientras que en el Chiari I se observa en menor porcentaje, pero con alteraciones significativas de la dinámica del LCR.
- Síntomas en la Infancia: Rápido aumento de la circunferencia de la cabeza o un tamaño extraordinariamente grande, vómitos, sueño, irritabilidad, desvío de los ojos hacia abajo (signo de "puesta de sol") y convulsiones.
- Síntomas en Niños Mayores y Adultos: Dolores de cabeza seguidos de vómito, náuseas, papiledema, visión borrosa, diplopía, problemas de equilibrio, coordinación deficiente, trastorno en el estilo de caminar, incontinencia urinaria, reducción o pérdida de desarrollo, letargo, somnolencia, irritabilidad y cambios en la personalidad o el conocimiento, incluida la pérdida de memoria.
- Tratamiento: Se trata con mayor frecuencia mediante la colocación quirúrgica de un sistema de derivación (válvula) que desvía el LCR a otra zona del cuerpo para su absorción.
- Complicaciones: Fallos mecánicos, infecciones, obstrucciones y la necesidad de prolongar o reemplazar el catéter.
Escoliosis
La asociación entre la escoliosis y la malformación de Chiari II se observa en el 50-70% de los pacientes, y en el Chiari I también existe una asociación, aunque menos conocida. La escoliosis es más frecuente en los casos con siringomielia asociada y se traduce por una afectación de la musculatura axial raquídea secundaria a una alteración progresiva de la motoneurona, con la consiguiente denervación de los músculos paravertebrales. La escoliosis no asociada a siringomielia suele ser dolorosa, con rápida progresión y una curvatura torácica izquierda.
Opciones de Tratamiento para la Malformación de Chiari
El tratamiento de la malformación de Chiari dependerá de varios factores, incluyendo el estado del paciente, la presencia de síntomas y la existencia de patologías asociadas como la siringomielia y la hidrocefalia.
Manejo Conservador
Si el estado del paciente es bueno y no hay síntomas ni patologías asociadas, el tratamiento de elección será conservador, siempre con revisiones regulares de control. Un tercio de los afectados nunca desarrollan síntomas y harán una vida completamente normal. En otros casos, los síntomas leves pueden resolverse con medicamentos para el dolor si el grado o la frecuencia no son severos.
Tratamiento Quirúrgico
Lo más común en esta enfermedad es que haya síntomas que mermen la calidad de vida de forma severa y gradual o que, sin haberlos aún, exista alguna patología asociada como siringomielia o hidrocefalia. En estos casos, la cirugía es la elección, pero siempre se tratará cada caso de forma individualizada. El primer paso en el tratamiento quirúrgico de la siringomielia es un diagnóstico preciso de su etiología para dirigir el tratamiento a la causa subyacente.
- Indicaciones: Solo se opera el Chiari sintomático o con progresión de siringomielia.
- Craniectomía o Descompresión Suboccipital: Es el tratamiento estándar que se realiza en la mayoría de los centros del mundo para este diagnóstico, con o sin siringomielia. La operación suele durar entre 3 y 5 horas, extrayendo una pequeña parte de hueso de la parte posterior del cráneo para dejar espacio al cerebelo y liberar presión. Lo más adecuado para un mayor éxito es abrir la duramadre y poner un parche sintético o con tejido del propio paciente antes de cerrar.
- Técnica Mínimamente Invasiva (Teoría del Filum Terminale): Desde 1993, con la publicación de la tesis doctoral del Dr. Miguel B. Royo Salvador, se ha propuesto una técnica alternativa.
- Tiempo quirúrgico de 45 minutos.
- Pocas horas de ingreso.
- Anestesia local.
- El propósito de la intervención quirúrgica es detener la evolución de la enfermedad y que ni la ectopia ni las lesiones aumenten ulteriormente.
- En los casos de siringomielia asociados con malformaciones cráneo-cervicales, el primer paso es realizar una descompresión de la unión cráneo-cervical.
- En los casos con respuesta insuficiente y persistiendo una cavidad intramedular grande, la derivación se debe realizar como un segundo procedimiento quirúrgico.
- Atención a la Hidrocefalia: Es aconsejable antes de cualquier cirugía cráneo-vertebral, ya que la propia hidrocefalia podría ser la causa de la siringomielia.
Complicaciones de la Cirugía
Dependiendo de la localización de la lesión, la misma intervención de descompresión puede causar déficit neurológico, con los siguientes porcentajes de incidencia:
- Hemiparesia (parálisis del hemicuerpo) de 0,5 a 2,1%.
- Alteración del campo visual de 0,2 a 1,4%.
- Trastorno del lenguaje de 0,4 a 1%.
- Déficit sensitivo de 0,3 a 1%.
Resultados y Seguimiento Postoperatorio
Después del procedimiento quirúrgico, generalmente hay poco cambio en la condición neurológica del paciente, ya que el objetivo es detener el progreso de la enfermedad, no recuperar los déficits neurológicos ya establecidos, de ahí la importancia de operar temprano. La fuerza del músculo y la atrofia muscular de los miembros superiores pueden mejorar ligeramente, pero no suele haber cambios en la pérdida de sensación. Puede conseguirse una mejoría moderada en las señales de las neuronas motoras superiores de los miembros inferiores.
La representación de imágenes de resonancia magnética normalmente muestran una desaparición de las cavidades de la siringomielia después del procedimiento quirúrgico. Los estudios de RM de control tienen que ser realizados cada seis meses para el seguimiento de la evolución de la enfermedad y poder actuar rápidamente en caso de reaparición.
Complicaciones de la Ectopia de las Amígdalas Cerebelosas
Las complicaciones de la ectopia de las amígdalas cerebelosas pueden depender del grado de tracción caudal o de la compresión asociada en el foro occipital.
- Muerte Súbita: Debido al control de las funciones cardio-respiratorias de la zona del tronco cerebral, donde se ubican las amígdalas cerebelosas y se desarrolla la compresión, los trastornos respiratorios durante el sueño pueden constituir un aspecto de la patología, pudiendo expresarse en apneas, fallos respiratorios o incluso en la muerte súbita del paciente.
Breve Historia de la Malformación de Chiari
- 1883: Descrita por primera vez por el cirujano anatomista John Cleland (1835-1925) de Pertshire, Escocia. Describió la elongación del vermis cerebeloso, el descenso del cerebelo y del IV ventrículo en un niño con hidrocefalia, encefalocele, espina bífida y mielosquisis.
- 1891 y 1896: Hans Chiari aportó nuevos casos y su clasificación de la malformación.
- 1907: Fue nominada por Schwalbe y Gredig como "malformación de Arnold-Chiari", reconociendo las contribuciones de Julius Arnold (1894).
tags: #ectopicas #migdalas #cerebelosas #ectopicas