El síndrome de deleción 22q11.2, también conocido como síndrome de DiGeorge, es una condición genética que resulta de la pérdida de una pequeña parte del cromosoma 22. Esta deleción puede afectar el desarrollo de múltiples sistemas del organismo, manifestándose con un fenotipo clínico variable que puede oscilar de moderado a grave.
Características Clínicas del Síndrome de Deleción 22q11.2
Las manifestaciones del síndrome de deleción 22q11.2 son diversas y pueden incluir:
Defectos Cardíacos Congénitos
Los defectos cardíacos congénitos son una de las características más frecuentes, presentándose en aproximadamente dos tercios de los casos. Estas malformaciones incluyen principalmente anomalías conotruncales como la comunicación interventricular, truncus arterioso, tetralogía de Fallot y la interrupción del arco aórtico. Las anomalías del arco aórtico y los anillos vasculares también son comunes.

Anomalías Craneofaciales y del Paladar
Más del 65% de los pacientes presentan anomalías palatinas, como la incompetencia velofaríngea, paladar hendido submucoso o úvula bífida, que pueden resultar en habla hipernasal y dificultades en la alimentación. El paladar hendido y el labio leporino abiertos son menos frecuentes. Los pacientes suelen mostrar rasgos faciales sutiles pero característicos, como ptosis, hipertelorismo, pliegues epicánticos, raíz nasal prominente, región malar plana y orejas pequeñas.
Disfunción Inmunológica
La inmunodeficiencia se debe a la aplasia o hipoplasia del timo, la glándula donde maduran las células T, esenciales para la respuesta inmune. Aunque la producción de células T puede mejorar con el tiempo, los pacientes presentan un mayor riesgo de infecciones faríngeas frecuentes y otras infecciones.
Hipocalcemia y Hipoparatiroidismo
La hipocalcemia, resultante del hipoparatiroidismo (funcionamiento reducido de las glándulas paratiroideas), es común en el período neonatal. Aunque a menudo remite, puede reaparecer a cualquier edad, manifestándose con niveles reducidos de calcio y elevados de fósforo en la sangre.
Problemas de Desarrollo y Cognitivos
En general, los pacientes experimentan dificultades de aprendizaje y retraso psicomotor. Existe un mayor riesgo de trastorno por déficit de atención e hiperactividad (TDAH) y trastornos del espectro autista en la infancia. La deleción puede causar problemas en el desarrollo mental, el aprendizaje, la conducta y el comportamiento social.
Otros Hallazgos Clínicos
Otros hallazgos adicionales pueden incluir anomalías gastrointestinales (malrotación intestinal, ano imperforado), pérdida auditiva, anomalías renales (agenesia renal), anomalías dentales (hipoplasia del esmalte) y anomalías esqueléticas (escoliosis, pie zambo). En un caso se describió hipoacusia bilateral neurosensorial severa y otitis de repetición, cornea grande y desnutrición leve.
Genética y Diagnóstico del Síndrome de Deleción 22q11.2
El síndrome se debe, en la mayoría de los casos, a una deleción de aproximadamente 3 millones de pares de bases en la región cromosómica 22q11.2, flanqueada por repeticiones de bajo número de copias. Esta deleción ocurre por recombinación meiótica no alélica durante la espermatogénesis u ovogénesis. Aproximadamente, en el 15% de los casos, la deleción se localiza dentro de la región crítica de DiGeorge de 3 Mb y su tamaño es variable. La mayoría de estas deleciones incluyen el gen TBX1, crucial para el desarrollo cardíaco, paratiroideo, tímico y facial.
La deleción surge de novo en aproximadamente el 90% de los casos. El riesgo de recurrencia entre hermanos en un caso de novo es del 2-3% debido al posible mosaicismo de la línea germinal parental de bajo grado. En un caso específico, se diagnosticó una microduplicación 22q11.2, lo que resalta la variabilidad genética dentro de esta región.
Diagnóstico Prenatal y Amniocentesis
El diagnóstico prenatal es posible en casos familiares o cuando se detectan anomalías asociadas mediante ecografía fetal. Las técnicas invasivas como la amniocentesis o la biopsia corial permiten obtener material genético del feto para detectar anomalías genéticas comunes, incluyendo la deleción 22q11.2.
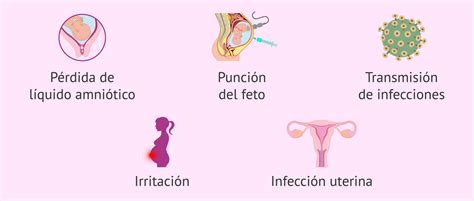
La amniocentesis generalmente se realiza entre las semanas 15 y 20 del embarazo para detectar alteraciones genéticas. Sin embargo, también puede realizarse entre la semana 32 y 36. Es importante considerar los riesgos asociados a este procedimiento, como la lesión con la aguja, el sangrado o la pérdida de líquido amniótico, la sensibilización al Rh, infecciones urinarias y un riesgo de aborto de aproximadamente el 1%.
Tests Prenatales No Invasivos
Como alternativa a las pruebas invasivas, existen tests prenatales no invasivos (TPNI) que analizan el ADN fetal libre en la sangre materna. Estos tests pueden evaluar el riesgo de trisomías (21, 18, 13), aneuploidías en los cromosomas X e Y, y detectar la probabilidad de la deleción 22q11.2 (síndrome de DiGeorge). Ejemplos de estos tests incluyen PrenatalSafe® Karyo y PrenatalSafe® Karyo Plus + GeneSafe® Complete, que ofrecen un análisis integral de cromosomas y genes.
Manejo y Pronóstico
El manejo del síndrome de deleción 22q11.2 es sintomático y requiere un enfoque multidisciplinar. Puede incluir cirugía cardíaca y/o palatina, alimentación nasogástrica, suplementación de calcio, terapia ocupacional, fisioterapia, logopedia, terapia educativa y conductual, así como tratamiento para trastornos psiquiátricos. Se recomienda vigilancia regular de los niveles de calcio, la función tiroidea y el recuento de células sanguíneas. La amigdalectomía no se recomienda a menos que lo indique un centro experto.
El pronóstico es variable y depende de la gravedad de las manifestaciones clínicas. La tasa de mortalidad infantil es relativamente baja (~4%), mientras que en adultos es mayor que en la población general. A pesar de las dificultades, la mayoría de las malformaciones congénitas y problemas médicos asociados pueden ser tratados, permitiendo a los pacientes llevar una vida plena con el apoyo adecuado.
SÍNDROME DE DIGEORGE
tags: #amniocentesis #y #sindrome #de #george