El Síndrome de Kallmann (SK), también conocido como Síndrome de Maestre-Kallmann-Morsier o hipogonadismo anósmico, es una enfermedad genética rara que afecta principalmente el desarrollo del aparato reproductor y el sentido del olfato. Es un trastorno que provoca la producción insuficiente de hormonas esenciales para el desarrollo sexual y otras funciones corporales. Esta afección se caracteriza por el hipogonadismo hipogonadotrópico (HH) y la anosmia (incapacidad total o hiposmia, que es la reducción de la capacidad para percibir olores).
El SK se debe a la incapacidad del hipotálamo para producir la hormona liberadora de gonadotropina (GnRH), esencial para el funcionamiento normal del sistema reproductivo. Si no se trata, las personas con síndrome de Kallmann permanecerán sexualmente subdesarrolladas e infértiles. Aunque es principalmente genético, ciertos factores ambientales pueden influir en su manifestación, pero existe evidencia limitada que vincule agentes infecciosos específicos o exposiciones ambientales directamente con su desarrollo.
El Síndrome de Kallmann afecta aproximadamente a 1 de cada 30.000 varones y a 1 de cada 120.000 mujeres, siendo más frecuente en hombres. El diagnóstico precoz y el inicio del tratamiento son fundamentales, ya que mejoran significativamente los resultados y la calidad de vida de las personas con esta condición.
Tipos de Síndrome de Kallmann
El síndrome de Kallmann se puede clasificar en dos tipos principales según las mutaciones genéticas:
- Tipo 1 (KS1): Este tipo es causado por mutaciones en el gen ANOS1 (anteriormente KAL1).
- Tipo 2 (KS2): Este tipo está asociado con mutaciones en otros genes como FGFR1, FGF8, PROKR2 y PROK2.
Ambos tipos comparten características comunes, pero pueden variar en gravedad y presentación.
Causas y Factores de Riesgo del Síndrome de Kallmann
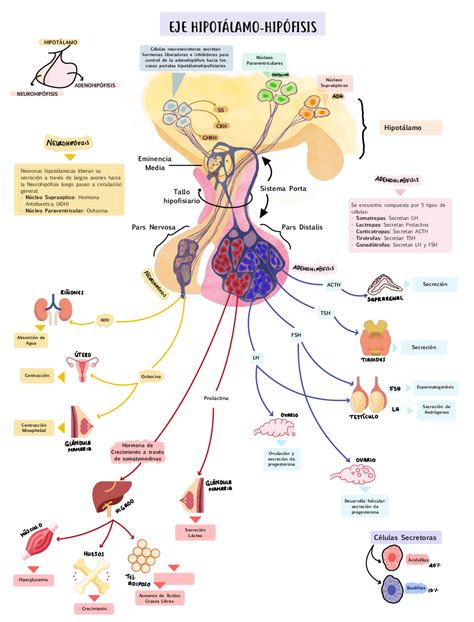
La causa principal del síndrome de Kallmann son mutaciones genéticas que afectan el desarrollo y la función de las neuronas productoras de GnRH. La principal causa de esta enfermedad es la deficiencia de la hormona GnRH debido a un fallo durante el desarrollo embrionario del individuo. Concretamente, se produce un defecto en la migración de las neuronas hipotalámicas que sintetizan la GnRH desde el epitelio olfatorio hasta la región hipotalámica del cerebro.
Además, también se produce un defecto en el desarrollo del sistema olfatorio y, por ello, las personas con síndrome de Kallmann sufren de anosmia o hiposmia. La GnRH es una de las hormonas que controlan el eje hipotálamo-hipofisario.
Las mutaciones genéticas pueden heredarse de forma:
- Herencia recesiva ligada al cromosoma X: El KS1 generalmente se hereda con un patrón recesivo ligado al cromosoma X, donde la mutación del gen ANOS1 (Xp22.32) se transmite de madre a hijo.
- Herencia autosómica dominante: La KS2 puede heredarse con un patrón autosómico dominante (genes FGFR1 (8p12), FGF8 (10q25-q26), CHD7 (8q12.2) y SOX10 (22q13.1)).
- Herencia autosómica recesiva: La KS2 también puede heredarse con un patrón autosómico recesivo (genes PROKR2 (20p12.3) y PROK2 (3p21.1)).
La mayoría de los casos de síndrome de Kallmann son esporádicos, es decir, ocurren al azar y no se heredan. Sin embargo, hay casos en los que la enfermedad se transmite de padres a hijos. Un nuevo concepto de herencia genética, denominado herencia oligogénica, sugiere que las mutaciones en varios genes pueden combinarse para causar la enfermedad, añadiendo otro nivel de complejidad a sus orígenes genéticos.
Síntomas del Síndrome de Kallmann
Los síntomas del síndrome de Kallmann pueden variar ampliamente, pero las características clínicas más comunes incluyen la ausencia de pubertad espontánea y la pérdida total o parcial del olfato. Desde el nacimiento, los varones pueden presentar rasgos como micropene o testículos no descendidos (criptorquidia), que indican signos precoces de la enfermedad. Sin embargo, los síntomas más evidentes suelen aparecer en la pubertad debido a la falta de maduración sexual. Dos rasgos distintivos del síndrome de Kallmann en los que se fijan los médicos a la hora de solicitar el diagnóstico son la falta de cambio físico/desarrollo sexual asociado a la pubertad y el deterioro o ausencia del sentido del olfato.
Síntomas en varones
Después de la pubertad y una vez llegados a la edad adulta, los hombres con síndrome de Kallmann tendrán un fenotipo relacionado con el hipogonadismo y la falta de la hormona testosterona:
- Crecimiento testicular escaso o nulo (volumen testicular pequeño).
- Disminución de la densidad ósea y masa muscular reducida.
- Disfunción eréctil.
- Disminución de la libido.
- Infertilidad (Oligospermia o azoospermia).
- Apariencia de adolescente (caracteres infantiles que persisten en la vida adulta).
Síntomas en mujeres
Los síntomas principales del síndrome de Kallmann en las mujeres son:
- Amenorrea primaria (ausencia de períodos menstruales).
- Desarrollo incompleto o ausente de las mamas al llegar a la pubertad.
- Esterilidad (anovulación debido a la falta de estrógenos).
Síntomas comunes a ambos sexos
- Deterioro o ausencia del sentido del olfato (anosmia o hiposmia) debido al desarrollo incorrecto de los bulbos olfativos.
- Retraso o ausencia de los signos de la pubertad.
- Disminución de la densidad ósea (aumento del riesgo de osteoporosis).
- Anomalías renales, como la agenesia renal unilateral.
- Retraso mental moderado.
- Déficit auditivo.
- Movimiento involuntario de las manos o de los globos oculares.
- Paladar hendido o labio leporino.
- Anomalías dentales.
- Anomalías esqueléticas, como dedos cortos.

Diagnóstico del Síndrome de Kallmann
El diagnóstico del síndrome de Kallmann puede ser complicado debido a su rareza y a su presentación variable. El proceso de diagnóstico suele implicar una combinación de evaluación clínica, pruebas hormonales y análisis genético. Para determinar si una persona sufre realmente el síndrome de Kallmann, existen diferentes métodos diagnósticos, que deberán hacerse una vez pasada la pubertad, después de los 15-16 años.
Evaluación clínica
Una evaluación clínica exhaustiva incluye una historia clínica detallada y un examen físico para evaluar el desarrollo puberal y otras características asociadas como la anosmia. Un endocrinólogo puede confirmar el diagnóstico mediante un examen clínico, análisis de sangre, pruebas de imagen y utilizando la herramienta de clasificación de Tanner, un método de diagnóstico establecido que se utiliza en el campo de la endocrinología para determinar la fase de desarrollo de determinados caracteres sexuales.
La evidencia clínica de hipogonadismo se manifiesta en la ausencia de caracteres sexuales secundarios, como la amenorrea, la disfunción eréctil o la falta de libido. También se valora cualitativa y cuantitativamente la percepción olfativa. Puede hacerse una resonancia magnética para ver los bulbos olfatorios.
Pruebas hormonales
Se realizan análisis hormonales para medir los niveles de hormonas clave, entre ellas:
- Gonadotropinas (LH y FSH): Los niveles bajos de hormona luteinizante (LH) y hormona folículo estimulante (FSH) son indicativos de hipogonadismo hipogonadotrópico.
- Hormonas sexuales (testosterona o estradiol): Los niveles bajos de hormonas sexuales respaldan aún más el diagnóstico.
Prueba genética
Las pruebas genéticas son fundamentales para confirmar el diagnóstico e identificar la mutación genética específica. Las técnicas como la secuenciación de nueva generación (NGS) pueden detectar mutaciones en genes relacionados con el SK. En algunos casos, pueden solicitarse pruebas genéticas para ayudar a diagnosticar las distintas formas de esta enfermedad.
Tratamiento del Síndrome de Kallmann
El tratamiento del síndrome de Kallmann tiene como objetivo principal inducir y mantener las características sexuales secundarias y la fertilidad. No existe cura para el síndrome de Kallmann, pero hay tratamientos que son eficaces para controlar la enfermedad. El tratamiento suele ser de por vida e incluye terapia hormonal de reemplazo, con otras opciones disponibles para quienes buscan la fertilidad. Sin tratamiento, la mayoría de los hombres y mujeres afectados no pueden tener hijos biológicos debido a la infertilidad.
Para inducir la pubertad y, más tarde, la fertilidad, se emplea la terapia hormonal sustitutiva, es decir, hormonas de forma exógena que restituyan los niveles normales. Por una parte, el vello corporal, la voz más grave, el desarrollo muscular y óseo en los hombres y, por otra parte, los pechos desarrollados, la menstruación, el vello púbico, etc. en las mujeres.
Con relación a la anosmia o hiposmia, característica también del síndrome de Kallmann, no existe todavía un tratamiento específico disponible.
Síndrome de Kallman
Tratamiento de la infertilidad
En el momento en que una persona afectada por el síndrome de Kallmann desee tener hijos, habrá que cambiar el tratamiento hormonal para poder restaurar la fertilidad. El empleo de hormonas de forma exógena puede activar la gametogénesis y, por lo tanto, la fertilidad en pacientes que padecen el síndrome de Kallmann.
Varón con síndrome de Kallmann
- Las inyecciones repetidas de GnRH promoverán la espermatogénesis y habrá espermatozoides a partir del tercer mes.
- También es posible probar con la administración de hCG (gonadotropina coriónica humana) seguida de FSH (hormona folículo estimulante), ya que aumentarán la concentración de testosterona intratesticular. En los hombres, las inyecciones de hCG pueden estimular los testículos para producir testosterona y esperma, mientras que combinadas con hCG, las inyecciones de FSH pueden mejorar la espermatogénesis.
Mujer con síndrome de Kallmann
- Se administrará medicación hormonal para provocar una estimulación ovárica suave y, a continuación, se programarán las relaciones sexuales. Esto es lo que se conoce como coito dirigido (probabilidad de embarazo 20-25%).
- Si hubiera un factor masculino leve como única alteración, se podría recurrir a un tratamiento de Inseminación Artificial (IA) (probabilidad de embarazo 15-20%).
- En caso de no conseguir el embarazo con estos tratamientos o si existen otras alteraciones más graves de la fertilidad, la pareja tendrá que recurrir a técnicas como la Fecundación In Vitro (FIV). En casos de FIV, teniendo en cuenta el componente hereditario, si se conoce la alteración genética, se podría plantear la posibilidad de hacer un estudio genético de los embriones (Diagnóstico Genético Preimplantacional, DGP) para seleccionar cuáles no tienen la alteración y evitar la transmisión de la enfermedad a la descendencia.
Los efectos del tratamiento de fertilidad pueden tardar hasta dos años en ser evidentes en el caso de los hombres, mientras que las mujeres pueden ovular tras solo una semana de tratamiento.
Prevención y Asesoramiento Genético
Actualmente, no se conoce ningún método para prevenir el síndrome de Kallmann debido a su naturaleza genética. Sin embargo, el asesoramiento genético puede brindar información valiosa a las familias afectadas y ayudar a comprender los riesgos de transmitir la enfermedad a la descendencia.
El asesoramiento genético implica evaluar los antecedentes familiares y los resultados de las pruebas genéticas para brindar información sobre los patrones hereditarios del síndrome de Kallmann y los riesgos de recurrencia. En aquellos casos en los que el hipogonadismo hipergonadotropo se asocie a un síndrome genético, sería recomendable realizar Diagnóstico Genético Preimplantacional en casos de fecundación in vitro.
tags: #sindrome #de #kallman #infertilidad