El síndrome de Horner congénito es una afección neurológica poco frecuente que se presenta al nacer y que afecta los nervios de la cara y los ojos. Este síndrome se caracteriza por una interrupción en la vía simpática oculosimpática en cualquier parte de su recorrido desde el sistema nervioso central hasta el globo ocular y sus anexos.
Generalmente, el síndrome de Horner afecta solo un lado de la cara y se manifiesta con un conjunto específico de síntomas que incluyen cambios en la apariencia de los ojos, caída de los párpados y, en los casos congénitos, una diferencia en el color del iris. Aunque la mayoría de los casos congénitos tienen una etiología benigna, es fundamental una evaluación exhaustiva para descartar enfermedades subyacentes graves.
Causas del Síndrome de Horner en recién nacidos
El síndrome de Horner congénito se produce debido a alteraciones o daños en la vía nerviosa simpática durante el desarrollo fetal o en el momento del parto. Las vías nerviosas afectadas se dividen en tres grupos de células nerviosas (neuronas):
- Neuronas de primer orden: Esta vía neuronal comienza en el hipotálamo posterolateral en la base del cerebro, pasa por el tronco encefálico y se extiende hasta la parte superior de la médula espinal (segmentos cervicotorácicos C8-T2).
- Neuronas de segundo orden: Esta vía neuronal se extiende desde la columna vertebral, cruza la parte superior del tórax y llega hasta el ganglio cervical superior a la altura de C3-C4.
- Neuronas de tercer orden: Esta vía neuronal se extiende a lo largo del costado del cuello, sigue la arteria carótida interna y externa, y llega hasta la piel del rostro y los músculos del iris y de los párpados, inervando las glándulas sudoríparas y los vasos sanguíneos de la cara.
Cualquier lesión o patología en el cerebro, tórax superior, cuello, cara u ojo puede ser responsable de la aparición del síndrome de Horner. Si bien las causas exactas no siempre están claras, se han identificado varios factores de riesgo y etiologías que pueden contribuir a la afección:
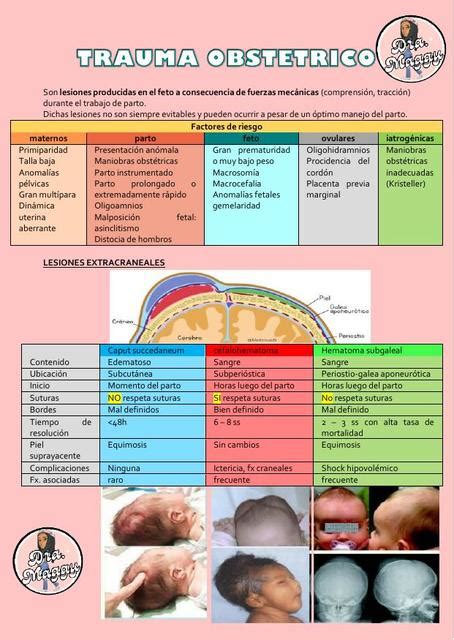
- Trauma obstétrico: Es la causa congénita más frecuente, suponiendo entre un 30% y un 50% de los casos. Comúnmente se asocia con lesiones del plexo braquial secundarias al parto.
- Causas idiopáticas: En muchos casos, no se encuentra ninguna patología responsable, y se considera entonces que es un Horner idiopático, de causa desconocida.
- Neuroblastoma: Aunque infrecuente, debe descartarse la posibilidad de un neuroblastoma torácico, que es la neoplasia implicada con mayor frecuencia y puede ser el primer (y a veces el único) signo de una enfermedad subyacente grave.
- Malformaciones vasculares: Como anomalías de la carótida.
- Síndrome de varicela congénito: En raras ocasiones, se han descrito otras causas como esta.
- Otras causas: Lesiones en nervios que controlan los músculos oculares y faciales, o alteraciones en la vía simpática durante el desarrollo fetal.
El síndrome de Horner puede estar presente al nacimiento o adquirirse posteriormente a cualquier edad, pero para considerarse congénito, debe manifestarse en las primeras 4 semanas de vida.
Síntomas y signos clínicos
El síndrome de Horner congénito se caracteriza por una tríada clásica de signos neurológicos que afectan un lado de la cara:
- Ptosis palpebral: Caída del párpado superior, que puede ser de 1 a 2 mm por afectación del músculo de Müller. En algunos casos, puede ser lo suficientemente grave como para dificultar la visión.
- Miosis: Contracción de la pupila, lo que origina una anisocoria (pupilas de tamaño desigual) de 1 a 1.5 mm, con la pupila afectada más pequeña. A pesar de esto, en la oscuridad se produce algún grado de dilatación pupilar debido a fuerzas pasivas. La anisocoria es máxima tras 5 segundos de oscuridad, cuando la pupila normal está dilatada al máximo y la pupila afectada aún no ha comenzado a dilatarse.
- Anhidrosis facial ipsilateral: Disminución o ausencia de sudoración en el lado afectado de la cara por denervación de fibras sudorales y vasoconstrictoras. En lactantes, la hemianhidrosis facial no siempre está presente, pero se pueden buscar equivalentes como el signo del Arlequín (hemianrojecimiento de la hemicara contralateral con el llanto) o la obstrucción nasal unilateral.
Además de la tríada clásica, se pueden presentar otros signos:
- Heterocromía de iris: Es un signo muy típico cuando el síndrome es congénito. El iris del ojo afectado tiene un color diferente, generalmente más claro, debido a la interrupción del desarrollo neurotrópico de los melanocitos del iris por la falta de estimulación de las fibras simpáticas postganglionares. En sujetos de ojos oscuros, el iris más claro corresponde a la pupila anormal; en los casos de ojos azules, el iris oscuro corresponde al lado afectado.
- Enoftalmos aparente: Hundimiento aparente del globo ocular.
- Hiperemia conjuntival y eritema facial: Enrojecimiento y dilatación de los vasos sanguíneos.
- Rubor hemifacial contralateral: Es un signo más fácil de cuantificar en el niño que la anhidrosis, su equivalente en adultos.
Los síntomas suelen manifestarse en la infancia o la niñez temprana. Es importante comunicar al médico los síntomas del niño, incluyendo cuándo empezaron y si han empeorado con el tiempo, así como cualquier afección médica previa o lesiones recientes.
Síndrome de Horner. Miosis unilateral y ligera ptosis palpebral
Diagnóstico del Síndrome de Horner
El diagnóstico del síndrome de Horner congénito es principalmente clínico y se basa en la observación de los signos y síntomas característicos. Es suficiente la existencia de miosis, con o sin ptosis, más uno de los siguientes signos: retraso de la dilatación pupilar en la oscuridad, heterocromía de iris, anhidrosis facial ipsilateral o causa evidente de interrupción de la vía oculosimpática.
Evaluaciones diagnósticas:
- Examen físico: El médico examinará los ojos y la cara del niño. La presencia de la tríada clásica suele ser suficiente para el diagnóstico.
- Pruebas farmacológicas:
- Test de cocaína: Históricamente utilizado, bloquea la recaptación de noradrenalina, pero la pupila afectada no se dilata debido a la interrupción simpática.
- Test de apraclonidina: Una alternativa más segura en niños, esta gota oftálmica causa dilatación de la pupila afectada y constricción de la pupila normal, lo que confirma el diagnóstico.
- Test de hidroxianfetamina: Permite establecer la localización de la lesión en la vía simpática (primera, segunda o tercera neurona), aunque su fiabilidad es escasa en el primer año de vida.
- Análisis de laboratorio:
- Determinaciones urinarias de ácido vanilmandélico (AVM) y ácido homovanílico (HVA): Se realizan para detectar tumores que secretan catecolaminas, como el neuroblastoma. Su normalidad no siempre descarta el neuroblastoma, ya que depende del volumen tumoral.
- Analítica sanguínea: Incluyendo hemograma, LDH (lactato deshidrogenasa), función renal y hepática.
- Pruebas de imagen: Son cruciales para encontrar la causa del daño nervioso y descartar una enfermedad subyacente grave, especialmente en casos de etiología desconocida o sin antecedentes de trauma obstétrico.
- Ecografía transfontanelar, cervical y abdominal: Permite visualizar posibles anomalías en estas regiones.
- Radiografía de tórax: Para detectar masas o tumores.
- Tomografía computarizada (TAC) helicoidal cervicotorácica con contraste: Puede ser una alternativa, especialmente en casos agudos o cuando la resonancia magnética no es posible, aunque tiene menor sensibilidad para lesiones vasculares.
- Resonancia magnética (RM) craneocervical (hasta T2-T3) con angiografía: Ofrece una buena sensibilidad para resaltar lesiones vasculares y otras anomalías en el trayecto nervioso implicado, con poca exposición a la radiación.
Es importante informar al médico sobre cualquier alergia al medio de contraste y asegurarse de que el niño no tenga objetos de metal durante una RM.
Tratamiento del Síndrome de Horner
El tratamiento del síndrome de Horner no requiere un tratamiento específico para la afección en sí, sino que se enfoca en abordar la causa subyacente. Si se identifica y se trata la causa, los signos y síntomas generalmente desaparecen.
Enfoques de tratamiento:
- Tratamiento de la causa subyacente: Si el síndrome de Horner es consecuencia de otra afección médica (como un tumor, un derrame cerebral o una lesión), el tratamiento se dirigirá a esa patología específica. Por ejemplo, en caso de un neuroblastoma, se seguirán los protocolos oncológicos correspondientes.
- Manejo de la ptosis palpebral:
- Intervención quirúrgica: Si la caída del párpado dificulta la visión o causa problemas estéticos significativos, se puede considerar la cirugía.
- Tratamiento tópico con fenilefrina: Una alternativa a la cirugía que puede ayudar a elevar el párpado.
- Terapia ocupacional: Puede ser útil si el niño tiene problemas para ver debido a la caída del párpado. Un terapeuta ocupacional puede ayudar a fortalecer los músculos del ojo afectado.
- Seguimiento oftalmológico: Un oftalmólogo (especialista en ojos) puede identificar y tratar los problemas de visión que se desarrollen.
En los casos de síndrome de Horner congénito idiopático (causa desconocida), que no presentan mayor problema que el estético debido a la caída del párpado, se realiza un seguimiento evolutivo del paciente. Es crucial que los padres participen activamente en la planificación del cuidado de su hijo, discutiendo las opciones de tratamiento con los médicos para decidir el cuidado que desean para él.