El Páncreas Ectópico
Introducción y Definición
El páncreas ectópico (PE) se define como la presencia de tejido pancreático fuera de su localización anatómica normal, careciendo de comunicación anatómica o vascular con el cuerpo principal del páncreas. Esta anomalía congénita fue descrita por primera vez en 1729 por Schultz y es la segunda anomalía congénita pancreática más frecuente, solo superada por el páncreas divisum.
Su incidencia es variable, oscilando entre el 0,25% y un 0,55% a 13% en estudios de autopsias. A menudo, su hallazgo es casual, detectándose en pruebas de imagen o durante intervenciones quirúrgicas realizadas por otros motivos.
Localización y Características Histológicas
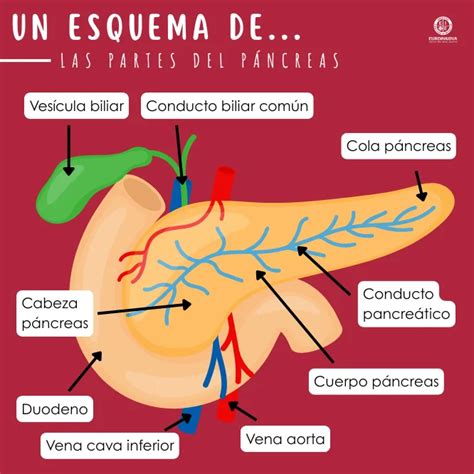
La localización más frecuente del páncreas ectópico es en el estómago, especialmente en la región prepilórica y sobre la curvatura mayor, donde se encuentra en el 25-38% de los casos o incluso hasta en el 70% según algunas series. Le siguen en frecuencia el duodeno, el yeyuno y el íleon.
Histológicamente, el tejido pancreático heterotópico puede presentar todos los elementos de una glándula pancreática normal (tipo I), incluyendo acinos, ductos e islotes de Langerhans. Sin embargo, también pueden encontrarse solo islotes (tipo IV, páncreas endocrino) o predominar nódulos de páncreas exocrino con formación de acinos y estructuras ductales.
En cuanto a la afectación de las capas del órgano donde se asienta, en el estómago, la localización más habitual suele ser la capa submucosa. No obstante, puede asentar en la muscular (17%) o en la serosa (10%).
Presentación Clínica y Síntomas
Aunque la mayoría de los casos de páncreas ectópico son asintomáticos, este tejido puede sufrir los mismos procesos patológicos que una glándula pancreática nativa. Aproximadamente el 40% de los casos son sintomáticos, con una presentación más frecuente en varones en torno a la quinta y sexta década de la vida.
Los síntomas más frecuentes suelen ser la epigastralgia (dolor en la parte superior del abdomen), dispepsia, sensación de plenitud y pirosis. Además, pueden manifestarse clínicamente con enfermedades pancreáticas como pancreatitis, o desarrollar neoplasias como carcinoma, tumor de células de los islotes y quistes. Los carcinomas que se originan en el páncreas ectópico suelen ser adenocarcinomas o carcinomas anaplásicos, aunque también se han descrito neoplasias mucinosas papilares intraductales, cistoadenocarcinomas o insulinomas.
Diagnóstico
Métodos Diagnósticos
El diagnóstico de páncreas ectópico es difícil por radiología, ya que el hallazgo radiológico no es específico, especialmente cuando el tejido ectópico se enmascara como otra lesión.
Las biopsias endoscópicas convencionales de la mucosa no suelen ser útiles debido a la profundidad de estas lesiones subepiteliales. La ecoendoscopia (EUS) es una herramienta valiosa, ya que permite determinar las características de la lesión, su origen en la pared gástrica y su patrón ecogénico y tamaño. Una lesión subepitelial hipoecoica, homogénea, originada en la tercera capa (submucosa) con una estructura ductal en su interior, es muy sugestiva de páncreas ectópico. Sin embargo, la EUS por sí sola no puede determinar de forma absoluta el tipo de lesión ni si es benigna o maligna, con una sensibilidad y especificidad de 64% y 80% respectivamente para diferenciar lesiones benignas de malignas.
La confirmación definitiva del diagnóstico se obtiene tras el estudio histopatológico, realizado después de la cirugía o de un estudio endoscópico más invasivo como la punción aspiración con aguja fina (PAAF) guiada por EUS, o la resección endoscópica.
Diagnóstico Diferencial
Entre los diagnósticos diferenciales del páncreas ectópico se pueden destacar el tumor del estroma gastrointestinal (GIST), el leiomioma, el linfoma y el bazo accesorio, los cuales también pueden presentarse como lesiones subepiteliales.
Casos Clínicos
Se ha documentado el caso de una mujer de 39 años remitida a consulta de Endocrinología por un cuadro clínico compatible con hipoglucemia. Tras las comidas, la paciente presentaba dolor y distensión abdominal, acompañados de temblor, sudoración y cefalea, con mejoría tras la ingesta de hidratos de carbono. En un episodio, se registró una glucemia capilar de 48 mg/dL. Tras un extenso estudio, que incluyó resonancia nuclear magnética (RNM) y ecoendoscopia con PAAF (inicialmente sugestiva de neoplasia neuroendocrina), se realizó una segunda endoscopia con toma de muestras para anatomía patológica. El resultado fue un acúmulo de tejido pancreático heterotópico/ectópico en la submucosa y mucosa gástrica. Dada la persistencia de los síntomas, se optó por la gastrectomía parcial, con resolución completa del cuadro. El análisis anatomopatológico de la pieza quirúrgica reveló una lesión nodular tumoral benigna (2,5 cm) de tejido pancreático heterotópico con acinos exocrinos, islotes de Langerhans y ductos epiteliales, afectando desde la submucosa hasta la subserosa con hiperplasia muscular.
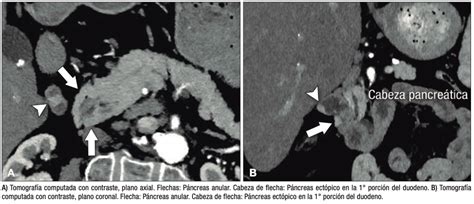
Otro caso reportado es el de un varón de 35 años que consultó por dolor abdominal súbito e intenso. La analítica mostró leucocitosis y proteína C reactiva elevada, mientras que una tomografía computarizada (TC) evidenció neumoperitoneo y líquido libre intraabdominal. Con el diagnóstico de perforación de víscera hueca, se realizó cirugía urgente. Durante la laparotomía, se suturó una perforación gástrica y, incidentalmente, se objetivó una masa firme de 5 cm en el yeyuno proximal. Se practicó una resección intestinal de la lesión, cuyo estudio histopatológico estableció el diagnóstico de páncreas ectópico.
Manejo y Tratamiento
El tratamiento del páncreas ectópico no está claramente establecido debido a que la degeneración maligna es infrecuente. Sin embargo, cuando se evidencia intraoperatoriamente, se recomienda la resección con márgenes libres para obtener un diagnóstico definitivo mediante estudio histológico.
Para lesiones subepiteliales gástricas menores de 3 centímetros, la endosonografía y la resección endoscópica de la mucosa (REM) han cambiado el manejo, permitiendo la resección en bloque en una sesión endoscópica, especialmente si la lesión se origina en las tres primeras capas (mucosa y submucosa). La técnica de resección endoscópica de la mucosa asistida por capuchón permite la resección en bloque de lesiones pequeñas para estudio citopatológico con baja tasa de complicaciones. Esta técnica no se recomienda para lesiones mayores de 15 mm o aquellas que se originan en la cuarta capa (muscular propia) debido al riesgo de perforación y sangrado.
Actualmente, la Asociación Americana de Gastroenterología (AGA) recomienda individualizar cada caso para el tratamiento óptimo de lesiones subepiteliales gástricas asintomáticas, incluyendo vigilancia periódica endoscópica o ecoendoscópica, o resección endoscópica o quirúrgica, y sugiere no dar seguimiento al páncreas ectópico gástrico asintomático.
Síndrome de Gorlin (Síndrome del Nevo Basocelular Nevoide)
Introducción y Epidemiología
El Síndrome de Gorlin (SG), también conocido como síndrome del carcinoma basocelular nevoide (OMIM: 109400), es una enfermedad genética rara de herencia autosómica dominante. Predispone al desarrollo de defectos del desarrollo y a múltiples neoplasias, destacando los carcinomas basocelulares (CBC) múltiples que aparecen a edades tempranas.
La prevalencia del SG varía según las series publicadas, estimándose entre 1/30827 y 1/256000. Se ha establecido una prevalencia mínima de 1/57000 habitantes, y se calcula que uno de cada 200 pacientes con uno o más CBC puede tener SG.
La esperanza de vida de los afectados por SG es de aproximadamente 73,4 años, lo cual es significativamente inferior a la de la población general. La causa más importante de muerte prematura en estos pacientes es el meduloblastoma.
Patogenia Molecular
El Síndrome de Gorlin es una enfermedad genética de herencia autosómica dominante, caracterizada por una alta penetrancia y expresividad variable. Se produce principalmente por la pérdida de heterocigosidad del gen supresor tumoral PTCH1, mapeado en el cromosoma 9q22.32.
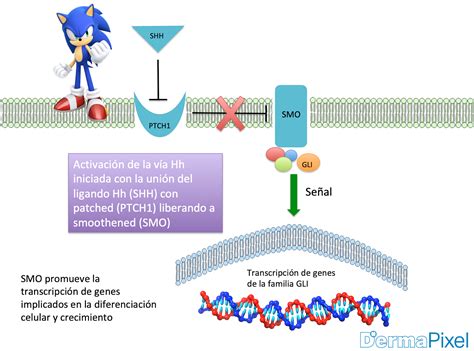
PTCH1 codifica para el receptor transmembrana PTCH1, que es un componente clave de la vía de señalización Sonic Hedgehog (SHH). Las mutaciones en este gen conllevan una sobreexpresión de la ruta SHH. La vía SHH, descrita por primera vez en Drosophila, es esencial durante el desarrollo embrionario, interviniendo en la polaridad tisular y la población de células madre. En mamíferos, está compuesta por cuatro elementos principales:
- Ligandos Hedgehog (LHH) de PTCH1: Sonic Hedgehog, Indian Hedgehog y Desert Hedgehog.
- El receptor PTCH1.
- La proteína transductora de señales smoothened (SMO).
- Los factores de transcripción Gli1, Gli2, Gli3.
Normalmente, PTCH1 inhibe constitutivamente la actividad de la proteína SMO. La unión de los LHH a PTCH1 suprime esta actividad inhibitoria, liberando a SMO para que se trasloque al cilio primario y active los factores de transcripción Gli. Las proteínas Gli promueven la transcripción de genes implicados en el aumento de la supervivencia celular y la mitosis. En vertebrados, GLI1 y GLI2 tienen función activadora, mientras que GLI3 impide la transcripción de los genes diana. Se ha demostrado una relación entre la vía SHH y otras rutas de señalización que modulan la patogenia del cáncer, como las del factor de crecimiento epidérmico, factor de crecimiento insulínico, factor de crecimiento transformante beta (TGF-β), mammalian Target of Rapamycin (mTOR)/S6K1, receptor proteína quinasa c 1, notch, wnt/β-catenina y fosfoinosítido-3-quinasa (PI3K/Akt).
PTCH1 puede estar mutado en el 50-85% de los pacientes con SG, siendo el 20-30% de estas mutaciones de novo. Con menor frecuencia, se encuentran mutaciones en otros genes de la vía SHH, como Suppressor of fused (SUFU) y PTCH2, o en el gen SMO o GLI. El gen alterado con mayor frecuencia después de PTCH1 es SUFU, y debe ser investigado en pacientes con un test genético negativo para PTCH1. Las mutaciones inactivadoras de SUFU se asocian con menor penetrancia, menor número de criterios diagnósticos mayores, mayor riesgo de meduloblastoma y ausencia de queratoquistes odontogénicos. Las mutaciones en PTCH2 son raras y suelen conferir un fenotipo más leve.
Los pacientes con SG nacen con una mutación heredada en uno de los alelos de PTCH1 (primer "hit"). Para que la enfermedad se manifieste, se requiere un segundo "hit" o evento, que corresponde a una mutación adquirida sobre el alelo sano del gen, a menudo por factores externos como la luz UV.
Manifestaciones Clínicas y Criterios Diagnósticos
Hasta la fecha, no se ha logrado establecer una correlación definitiva entre el fenotipo y el genotipo en pacientes con SG. Diversas clasificaciones de criterios diagnósticos han sido descritas, incluyendo manifestaciones cutáneas y extracutáneas. Un documento de consenso de 2011 incluyó por primera vez el estudio molecular. Para el diagnóstico de SG, se requieren dos criterios mayores, o un criterio mayor y dos menores, o un criterio mayor y la confirmación molecular.
Manifestaciones Cutáneas
- Carcinomas basocelulares (CBC): Pueden aparecer a edades tempranas y ser múltiples, afectando tanto a áreas fotoexpuestas como no fotoexpuestas, con un ligero predominio en las expuestas. Las localizaciones más frecuentes varían entre sexos.
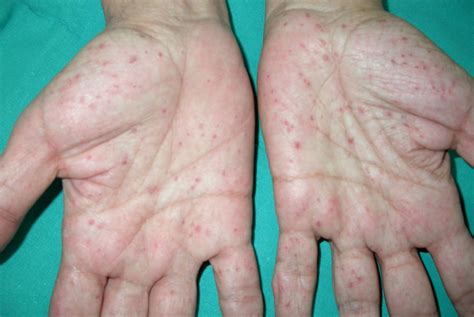
- Pits palmoplantares: Son una manifestación frecuente (70-87% de los enfermos). Son depresiones puntiformes de 2-3mm de diámetro en palmas y plantas, que suelen aparecer antes de los 15 años. Histológicamente, son áreas de hipoqueratosis con hipogranulosis variable.
- Otras manifestaciones cutáneas: Nevus melanocíticos múltiples, quistes de millium en párpado inferior y frente, y quistes epidermoides en tronco.
Manifestaciones Extracutáneas
Anomalías en el Desarrollo
- Queratoquistes mandibulares u odontogénicos: Presentes en el 74-90% de los pacientes, pueden iniciarse a los 4-5 años y aparecen antes de los 20 años en el 75% de los casos. Suelen ser asintomáticos, múltiples y bilaterales en la mandíbula.
- Manifestaciones no tumorales del sistema nervioso central: La calcificación de la hoz del cerebro es la más frecuente (65-79%), detectable a partir de la adolescencia. Otras alteraciones raras incluyen la calcificación de la tienda del cerebelo.
- Anomalías faciales: Macrocefalia relativa e hipertelorismo, a menudo asociado a telecanto. También se ha descrito abombamiento de huesos frontales, biparietales o temporales.
- Alteraciones en vértebras, costillas y escápula: Entre el 38-49% presentan anomalías costales, siendo las costillas bífidas las más comunes. Las malformaciones congénitas vertebrales (escoliosis, proceso espinoso bífido) son frecuentes. La enfermedad de Sprengel (elevación congénita de la escápula) se detecta en el 11-22% de los pacientes.
- Alteraciones en miembros: Radiotransparencias en forma de llama en radiografías de manos (30%), defectos de modelaje en manos y pies, sindactilia, polidactilia y acortamiento del quinto metacarpiano.
- Alteraciones oculares y auditivas: Además del hipertelorismo, se ha notificado exoftalmos, nistagmo rotatorio, estrabismo interno, cataratas congénitas, coloboma del iris y coroides, y microftalmia. Anomalías otológicas incluyen otosclerosis, sordera de conducción y angulamiento posterior de las orejas.
Tumores no Cutáneos
- Meduloblastoma: Es el tumor maligno cerebral más frecuente en la población pediátrica y se desarrolla en el 1-5% de los pacientes con SG, a menudo como primera manifestación. Su pronóstico es intermedio, con tasas de supervivencia global entre 60-80%.
- Fibromas cardiacos y ováricos: Los fibromas cardiacos son infrecuentes y pueden causar arritmias o insuficiencia cardiaca. Los fibromas ováricos son generalmente incidentales y asintomáticos, aunque pueden causar problemas de fertilidad.
- Otros tumores: Meningiomas (hasta 5%), astrocitoma, craneofaringioma, oligodendroglioma, y tumores sólidos y hematológicos más raros como el rabdomioma fetal.
Pruebas Diagnósticas y Recomendaciones para el Seguimiento
Es fundamental minimizar la exposición a radiaciones ionizantes, por lo que se aconseja el uso de resonancia magnética o ecografía cuando sea posible. Las recomendaciones para el diagnóstico y seguimiento de los pacientes con SG incluyen:
- Estudio genético: Constituye un criterio mayor. Se recomienda en el diagnóstico prenatal si la mutación familiar es conocida, para confirmación en pacientes con signos incompletos, o como test predictivo en individuos con riesgo y familiares afectados. Se logra demostrar mutación en PTCH1 en el 60-80% de los casos.
- Radiografías simples: De parrilla costal y columna vertebral para descartar anomalías congénitas. Las radiografías de manos y pies pueden detectar cambios en la infancia. La radiografía craneal para la calcificación de la hoz cerebral es útil en adultos a partir de la adolescencia.
- Ortopantomografía digital: Recomendada anualmente desde que el niño colabore (a partir de los 4 o 8 años) hasta la aparición del primer queratoquiste, y cada 6 meses hasta los 21 años. En adultos, puede repetirse anualmente si hay síntomas.
Síndrome de Gorlin
Tratamiento
El tratamiento dermatológico del SG puede ser complicado debido al alto número y extensión de los carcinomas basocelulares. Recientemente, se han desarrollado nuevos fármacos que inhiben la vía Sonic Hedgehog, como el vismodegib, indicado para el tratamiento de CBC metastásico, recurrente o localmente avanzado. Aunque prometedores, su eficacia está limitada por los efectos secundarios y el desarrollo de resistencias.
El tratamiento del meduloblastoma, un tumor cerebral frecuente en niños con SG, es la cirugía combinada con radioterapia y quimioterapia. Sin embargo, en pacientes con SG, el tratamiento radioterápico puede inducir la aparición de CBC y otros tumores cerebrales en el área de radiación, por lo que es crucial una identificación temprana para optimizar el tratamiento adyuvante.