Introducción a los Programas de Cribado Neonatal
Los programas de cribado neonatal (PCN) son esenciales para la prevención de enfermedades en la infancia. A pesar de las variaciones en su diseño, organización y recursos en cada Comunidad Autónoma, todos son reconocidos como pilares fundamentales en los sistemas sanitarios para la prevención en Salud Pública. En España, se recomienda el cribado neonatal en muestras de sangre impregnada en papel para detectar, como mínimo, ocho entidades: hipotiroidismo congénito (HC), fenilcetonuria (PKU), deficiencia de acil CoA deshidrogenasa de ácidos grasos de cadena media (MACDD), deficiencia de 3-hidroxi acil CoA deshidrogenasa de ácidos grasos de cadena larga (LCHADD), aciduria glutárica tipo I (AG-1), deficiencia de biotinidasa (BTD), fibrosis quística (FQ) y anemia de células falciformes (AF).
Los orígenes del cribado neonatal se remontan a la década de 1960 con el desarrollo de la prueba bacteriológica de Guthrie, capaz de detectar niveles elevados de fenilalanina en una gota de sangre seca. Con el tiempo, la técnica de análisis y las metodologías disponibles han evolucionado, permitiendo coexistir hasta hace poco programas de muestra única de sangre seca y otros de muestra doble. La incorporación de la espectrometría de masas en tándem (MS/MS) en los laboratorios de cribado ha revolucionado el campo, posibilitando la detección de un número significativamente mayor de enfermedades, lo que ha dado lugar al denominado cribado metabólico expandido.
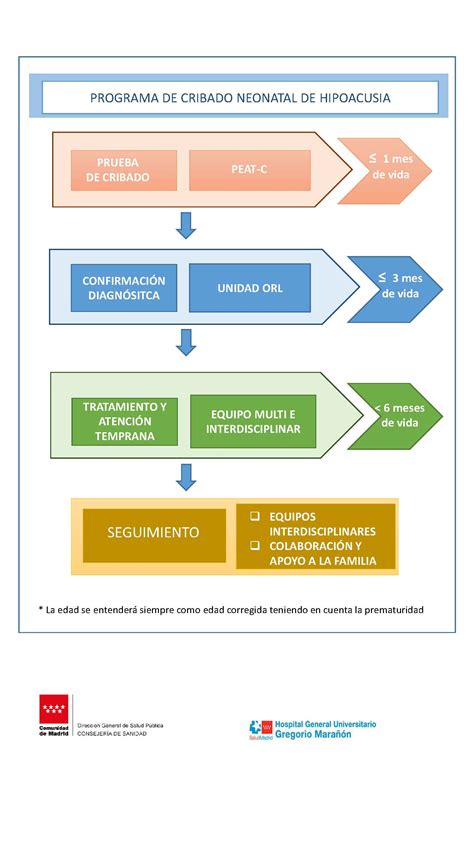
Objetivos y Metodología del Cribado Neonatal
El propósito principal de los análisis de cribado neonatal es identificar a los recién nacidos (RN) presuntamente positivos y clasificarlos según la probabilidad de padecer un trastorno específico, minimizando al mismo tiempo los resultados falsos positivos. Esto se logra estableciendo puntos de corte arbitrarios para los analitos o marcadores cuantificados. Es crucial entender que los PCN no son procedimientos de diagnóstico definitivo, sino que identifican grupos de alto riesgo que requieren estudios diagnósticos posteriores. Por ello, los PCN deben integrarse con unidades clínicas y de laboratorio especializadas en el diagnóstico y tratamiento de las enfermedades cribadas.
El beneficio más importante de un PCN es la prevención de discapacidades asociadas a las enfermedades. Los programas se organizan de diversas maneras para adaptarse a las distintas estructuras sanitarias, buscando siempre la máxima calidad analítica y cobertura. La estrategia debe planificarse para alcanzar una cobertura del 100% de los RN y el tratamiento temprano del 100% de los casos detectados.
Criterios para la Inclusión de Trastornos en el Cribado Masivo
Clásicamente, se definieron criterios para incluir un trastorno en el cribado masivo:
- La enfermedad debe cursar con morbilidad grave (retraso mental) o mortalidad si no se diagnostica en el período neonatal.
- Debe existir un tratamiento eficaz disponible.
- La frecuencia de la enfermedad debe ser relativamente elevada (al menos 1 de cada 10.000-15.000 RN).
- Debe existir un método analítico de cribado rápido, fiable y de bajo coste (Wilson & Jungner, 1968).
Aunque estos criterios siguen siendo vigentes, la tecnología MS/MS ha permitido considerar la inclusión de enfermedades con muy baja prevalencia (< 1:50.000 RN) si se detectan con la misma prueba analítica. En este contexto, la Academia Americana de Pediatría (AAP) y la Asociación Americana de Genética Médica (ACMG) emitieron en 2005 recomendaciones para EE.UU. sobre los trastornos a cribar, lo que ha llevado en algunas regiones a cribar más de 50 condiciones genéticas, a pesar de la posible falta de familiaridad de los profesionales de Atención Primaria con muchas de ellas.
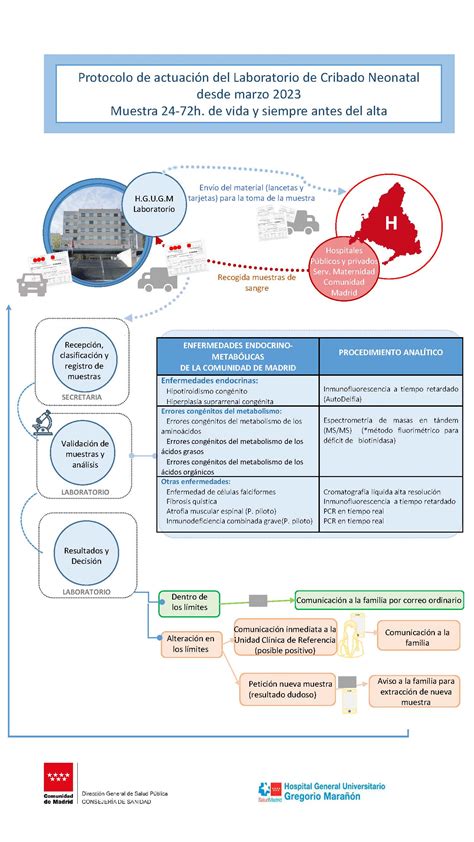
Toma y Manejo de Muestras en el Cribado Neonatal
Las estrategias de obtención de muestras biológicas pueden variar, pero generalmente se recomienda una única extracción de sangre capilar a partir de las 48 horas de vida del RN, tras una ingesta de 24 horas de alimento. La extracción debe ser realizada exclusivamente por personal sanitario, utilizando la punción del talón como procedimiento habitual. Para minimizar el dolor, se sugiere administrar lactancia materna, soluciones de glucosa o chupetes con sacarosa durante el procedimiento. La punción debe realizarse con lanceta estéril (< 2,4 mm) en la porción plantar del talón, para evitar daños osteo-articulares. Se recomienda masajear el pie para aumentar el flujo sanguíneo y desinfectar con alcohol de 70º, evitando derivados yodados. Tras la punción, se limpia la primera gota de sangre y se deja formar una nueva gota grande que caiga sobre el papel absorbente, asegurando que llene el círculo por completo con una sola aplicación.
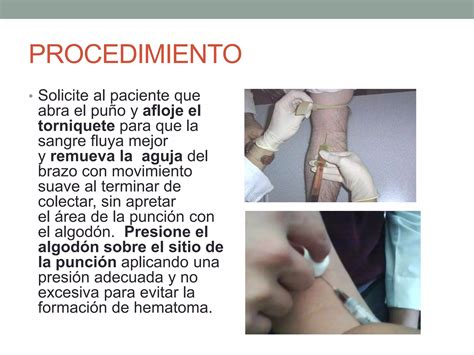
La sangre debe aplicarse en un solo lado del papel, verificando que haya traspasado uniformemente. Para una muestra óptima, las manchas de sangre deben contener al menos 75 μl (aproximadamente 13 mm de diámetro). Deben secarse en una superficie horizontal, limpia y seca, a temperatura ambiente, evitando la luz solar directa, durante al menos una hora. Las muestras deben enviarse al laboratorio lo antes posible, preferiblemente dentro de las 24-48 horas siguientes a la extracción. Es importante evitar el contacto entre muestras y no tocar ni manchar las gotas de sangre con agua, desinfectantes, jabones o alcohol para prevenir contaminaciones e interferencias analíticas.
Otras Muestras y Situaciones Clínicas Relevantes
También es posible realizar estudios de cribado a partir de muestras de orina en papel de filtro, utilizando técnicas como MS/MS o cromatografía de gases y espectrometría de masas (GC/MS). Estas muestras pueden servir para la confirmación diagnóstica, complementar la información de la muestra de sangre o ampliar las posibilidades diagnósticas. En algunas regiones de Canadá y países asiáticos, ya se han iniciado PCN con muestras de orina.
Se deben considerar situaciones clínicas especiales:
- RN con patología grave al nacimiento: se recomienda tomar una muestra al ingreso (idealmente antes de tratamiento antibiótico o nutrición parenteral) y repetirla a las 48-72 horas de vida.
- Transfusiones previas a la toma de muestra: obligan a una toma de muestra 48-72 horas después de la transfusión y repetirla a los 120 días.
- RN alimentados con nutrición parenteral: se debe realizar una toma de muestra 48-72 horas después de iniciar la alimentación enteral, independientemente de la edad.
Enfermedades Metabólicas Hereditarias (EMH) Incluidas en el Cribado
Hipotiroidismo Congénito (HC)
El HC tiene una importancia capital debido a su potencial repercusión en el desarrollo intelectual, ya que las hormonas tiroideas son esenciales para el desarrollo cerebral, siendo la causa más frecuente de retraso mental prevenible. Su frecuencia (1/3.500 RN vivos) justifica el programa de cribado. El diagnóstico precoz y el inicio del tratamiento dentro del primer mes de vida se asocian a coeficientes de inteligencia normales, sin problemas de aprendizaje y con crecimiento satisfactorio. Existen formas transitorias de HC relacionadas con la prematuridad, el uso de compuestos yodados o el consumo materno de ciertos medicamentos.
El cribado sistemático se basa en la determinación del nivel de TSH en sangre del talón, depositada en papel de filtro. Niveles elevados de TSH (≥ 10µUI/ml) indican hipotiroidismo primario. El estudio de confirmación se realiza midiendo los niveles séricos de T4 libre, que habitualmente está descendido, aunque puede ser normal en algunos casos. Situaciones de alteración hormonal intermedia incluyen hipertirotropinemia (TSH elevada con T4 normal) o hipotiroxinemia (TSH normal con T4 baja).
Fenilcetonuria (PKU)
La PKU es un paradigma de las EMH incluidas en los PCN. Es una enfermedad del metabolismo de la fenilalanina (Phe) con herencia autosómica recesiva. Su prevalencia varía entre 1/12.000 y 1/18.000 RN, según se consideren las formas clásicas o moderadas (hiperfenilalaninemias, HFA). El cribado se realiza midiendo por MS/MS los niveles de Phe y tirosina (Tyr) en sangre extraída a las 48-72 horas de vida del RN. La alta sensibilidad de esta tecnología ha permitido adelantar la toma de muestras en comparación con métodos antiguos que requerían 4-5 días de ingesta alimentaria.
Trastornos de la Beta-Oxidación
Los trastornos de la beta-oxidación mitocondrial son EMH de los ácidos grasos, debidos a mutaciones en MCADD o LCHADD, con herencia autosómica recesiva. Su prevalencia es muy variable (1/9.000 a 1/100.000 RN). Estos fallos provocan la acumulación de ácidos grasos y sus derivados (acilcarnitinas, acilglicinas) en sangre y orina, lo que lleva a problemas fisiopatológicos, defecto energético y/o acumulación de moléculas tóxicas. La clínica aparece en situaciones de descompensación metabólica, con aumento de las necesidades energéticas o disminución del aporte alimenticio.
Aciduria Glutárica Tipo I (AG-1)
La AG-1 es una enfermedad del metabolismo de los ácidos orgánicos causada por un defecto en la enzima GCDH (glutaril-CoA-deshidrogenasa), con herencia autosómica recesiva. Su prevalencia en España es de 1/85.000 RN. Esta enzima metaboliza los aminoácidos lisina (Lys) y triptófano (Trp). Su deficiencia provoca la acumulación de compuestos tóxicos para el sistema nervioso. Al nacimiento, el bebé puede no presentar problemas, pero la acumulación de derivados tóxicos comienza con la alimentación. Procesos infecciosos, fiebre o ayuno prolongado pueden desencadenar crisis encefalopáticas (convulsiones, irritabilidad, hipotonía).
Deficiencia de Biotinidasa (BTD)
Recientemente se ha incorporado el cribado de la deficiencia de biotinidasa (BTD), una forma de aparición tardía del déficit múltiple de carboxilasas. Sin tratamiento, se caracteriza por convulsiones, dificultad respiratoria, hipotonía, erupciones cutáneas, alopecia, pérdida de audición y retraso en el desarrollo. La prevalencia clínica del BTD se estima en 1/61.000 RN, con una frecuencia de portadores de aproximadamente 1/120. Los síntomas suelen aparecer en los primeros meses de vida, pero pueden manifestarse posteriormente. Individuos con déficit profundo sin tratar (<10% de actividad normal) presentan hallazgos clínicos variables. Metabólicamente, pueden mostrar acidosis cetoláctica, acidemia orgánica e hiperamonemia leve. Individuos con déficit parcial (<30% de actividad normal) pueden ser asintomáticos, pero desarrollar síntomas durante periodos de estrés. El tratamiento primario consiste en suplementos de biotina oral, que mejora los síntomas y previene su aparición. Una vez desarrollados algunos rasgos (atrofia óptica, pérdida de audición, retraso del desarrollo), pueden no ser reversibles. El tratamiento con biotina es de por vida y no se conocen efectos adversos serios.
Escuela Madrileña de Salud - Cribado Neonatal
Manejo de Resultados Alterados y Falsos Positivos
El manejo de los resultados del cribado neonatal se divide en dos categorías principales:
- Resultados alterados, altamente sugestivos de una alteración metabólica grave: Se debe contactar a la familia de inmediato, idealmente a través de un profesional con experiencia en el manejo de enfermedades metabólicas. Este profesional explicará a los padres el significado del resultado y los pasos a seguir para confirmar o excluir el diagnóstico.
- Resultados fuera del rango normal, pero no tan alterados como para indicar una enfermedad: Estos casos sospechosos requieren una repetición del test para distinguir entre un verdadero positivo y una anomalía transitoria debida a la inmadurez del RN. La solicitud de una nueva muestra suele realizarse mediante carta del laboratorio de cribado, explicando la anormalidad inicial y la necesidad de una muestra adicional. Se aconseja a los padres acudir a su pediatra, responsable de asegurar la repetición del test y de proporcionar información adicional.
La Federación Española de Enfermos Metabólicos Hereditarios (Feemh) ha solicitado la inclusión obligatoria de ciertas patologías en el cribado neonatal universal. Las aminoacidopatías, caracterizadas por el incorrecto metabolismo de aminoácidos y la acumulación tóxica de metabolitos, pueden afectar al sistema nervioso, hígado, riñones y visión. Los afectados requieren dietas bajas en proteínas para prevenir daño cerebral. La detección precoz es crucial, y aunque el cribado obligatorio en España cubre algunas aminoacidopatías como la fenilcetonuria y la aciduria glutárica I, existe una demanda de homogeneización y ampliación a otras como la tirosinemia, la enfermedad con olor a jarabe de arce y la aciduria (defecto del ciclo de la urea).
El diagnóstico temprano divide a los pacientes en dos grupos: aquellos sin secuelas neurológicas graves que llevan una vida normal, y los grandes dependientes con importantes secuelas cognitivas y motoras por no haber sido diagnosticados en la infancia. Un estudio de la Feemh reveló que la mitad de los cuidadores han tenido que dejar su trabajo, el 75% está en desacuerdo con el sistema de evaluación de discapacidad por ser arbitrario y depender de la comunidad autónoma, y el 75% reporta problemas de inclusión laboral.
El estudio también destacó la necesidad de crear la especialidad médica de metabolismo en adultos, ya que muchos pacientes continúan siendo tratados por pediatras metabólicos por falta de especialistas en la edad adulta. La capacidad de identificar de forma presintomática a los RN con riesgo de trastornos endocrinos, hematológicos y metabólicos ha salvado miles de vidas y ha generado ahorros significativos en el sistema sanitario desde la introducción de la prueba de Guthrie para la fenilcetonuria.
Interferencias Analíticas y Mejora de la Especificidad
Aunque las pruebas de cribado neonatal buscan alta sensibilidad para no pasar por alto bebés con trastornos, algunas pueden tener una especificidad diagnóstica inferior, resultando en un número de falsos positivos. Estos falsos positivos pueden causar ansiedad en los padres y someter a los bebés a pruebas médicas innecesarias, con el consiguiente gasto. Para minimizar esto, se utilizan pruebas de cribado de segundo nivel, que tienen un mayor grado de especificidad diagnóstica.
Una prueba de cribado primario puede ser un sistema de análisis de inyección de flujo basado en espectrometría de masas (FIA-MS/MS), un inmunoensayo u otra plataforma tecnológica. Muchas pruebas primarias tienen un alto valor predictivo positivo, incluso sin una prueba de segundo nivel. Sin embargo, para otros analitos, un resultado positivo del cribado primario se asocia con menor confianza, existiendo una probabilidad razonable de que no se diagnostique la enfermedad.
Ejemplos de Interferencias y Soluciones
- Detección de especies isobáricas de leucina por FIA-MS/MS: La ausencia de separación por LC de la leucina, isoleucina y alloisoleucina, y la interferencia isobárica de la hidroxiprolina, significa que un resultado elevado de "leucinas" no siempre equivale al diagnóstico de la enfermedad de la orina de jarabe de arce. La prueba ideal de segundo nivel es una técnica basada en separación que resuelva y cuantifique todas las especies isobáricas de leucina.
- Aumento de propionilcarnitina elevada: En el cribado primario de acidemias propiónica o metilmalónica, un aumento de propionilcarnitina puede también encontrarse en bebés de madres con deficiencia grave de vitamina B12. Una medición de segundo nivel de ácidos orgánicos mediante LC-MS/MS puede determinar si el patrón de metabolitos sugiere una enfermedad o una deficiencia vitamínica materna.
- Mediciones por inmunoensayo de 17-hidroxiprogesterona (17-OHP) en el cribado de hiperplasia suprarrenal congénita (HSC): La prematuridad, bajo peso al nacer y sufrimiento neonatal se asocian a elevaciones transitorias de 17-OHP y otras hormonas esteroides relacionadas, que pueden presentar reacciones cruzadas con los anticuerpos del inmunoensayo. El análisis de esteroides suprarrenales por LC-MS/MS permite distinguir la 17-OHP de otras hormonas no asociadas a la HAC. La introducción de una prueba de segundo nivel basada en LC-MS/MS en la Clínica Mayo redujo significativamente los costes asociados al seguimiento de bebés no afectados.
tags: #interferencias #teraputicas #hplc #aminoacidos #cribado #neonatal