El hiperinsulinismo congénito (HIC) representa uno de los trastornos metabólicos más graves relacionados con el metabolismo de la glucosa en neonatos. Esta patología se caracteriza por una alteración en la secreción de insulina, lo que desencadena episodios recurrentes y graves de hipoglucemia. Dada la vulnerabilidad del cerebro neonatal a la privación de glucosa, la confirmación diagnóstica y el inicio de un tratamiento precoz son prioritarios para prevenir secuelas neurológicas permanentes.
Comprensión de la Hipoglucemia Neonatal
La hipoglucemia es una de las alteraciones metabólicas más frecuentes en el neonato, resultando de un desequilibrio entre el aporte de glucosa y su utilización. Durante las primeras 24-48 horas de vida, en los recién nacidos normales que transitan de la vida intrauterina a la extrauterina, las concentraciones de glucosa en plasma (GP) suelen ser menores que en etapas posteriores de la vida.
Transición Metabólica en la Vida Extrauterina
El período posnatal inmediato implica cambios dramáticos en múltiples sistemas. El mantenimiento de la homeostasis de la glucosa depende de ajustes en los sistemas endocrinos y metabólicos, permitiendo pasar de un aporte continuo de glucosa fetal a una fase con períodos de ayuno e ingesta. En la vida intrauterina, la provisión de glucosa al feto es estable a través del aporte placentario, lo que evita la necesidad de gluconeogénesis activa. La glucosa no utilizada se almacena en el hígado fetal en forma de glucógeno.
Con la sección del cordón umbilical, el aporte continuo de glucosa se interrumpe abruptamente, provocando una caída de la glucemia que alcanza su punto más bajo durante las primeras dos horas de vida. En el recién nacido a término con crecimiento intrauterino adecuado, se produce un aumento progresivo posterior. La recuperación de los niveles de glucosa depende de mecanismos contrarreguladores, activados tanto por el proceso del parto como por la disminución de la glucemia. Las catecolaminas aumentan dramáticamente, estimulando la liberación de hormona del crecimiento (GH) y glucagón. El incremento de catecolaminas y glucagón activa la glucogenólisis hepática, mientras que los picos de GH y cortisol promueven la gluconeogénesis. Los niveles de insulina descienden inicialmente y permanecen bajos durante varios días, facilitando la gluconeogénesis y la movilización del glucógeno.
Los depósitos hepáticos de glucógeno son limitados y disminuyen significativamente en las primeras 12 horas de vida. Por ello, el mantenimiento de la normoglucemia dependerá del aporte exógeno de nutrientes y/o de la gluconeogénesis endógena, que se inicia a las 2-3 horas del nacimiento y alcanza su máximo a las 12 horas. Para garantizar el acceso a sustratos alternativos para el cerebro durante este período de glucosa baja, se observa un aumento de cuerpos cetónicos.
Definición y Síntomas de Hipoglucemia Clínica
La hipoglucemia clínica se define como una concentración de GP suficientemente baja como para causar síntomas o signos de alteración de la función cerebral. En niños capaces de comunicar síntomas, se recomienda la evaluación y el manejo solo de aquellos en los que se documenta la tríada de Whipple: síntomas compatibles con hipoglucemia, concentración de GP baja documentada y alivio de los síntomas al restaurar la GP a la normalidad. En lactantes y niños pequeños que no pueden comunicar síntomas de manera fiable, se sugiere la evaluación cuando las concentraciones de GP documentadas por análisis de laboratorio de calidad están por debajo del umbral normal para las respuestas neurogénicas (<60 mg/dl [3,3 mmol/L]).
Los síntomas de la hipoglucemia reflejan las respuestas del cerebro a la privación de glucosa. Los síntomas neurogénicos (autonómicos) incluyen palpitaciones, temblor, ansiedad, sudoración, hambre y parestesias. Los signos y síntomas neuroglucopénicos, como confusión, coma y convulsiones, son causados por la disfunción cerebral debido a un suministro deficiente de glucosa. El cerebro adulto representa más de la mitad del consumo total de glucosa. Debido al tamaño desproporcionadamente mayor del cerebro en relación con la masa corporal, los niños y lactantes tienen una tasa de utilización de glucosa 2 a 3 veces más alta por kilogramo de peso corporal en comparación con los adultos.
Hiperinsulinismo Congénito (HIC): Una Causa Mayor de Hipoglucemia Persistente
Definición y Epidemiología
El hiperinsulinismo congénito (HIC) es la causa más común de hipoglucemia persistente, definida como valores menores a 40 mg/dl, en la etapa neonatal y en la infancia. Se calcula una incidencia global de 1 cada 30 000-50 000 nacidos vivos, llegando a ser de 1 cada 2500 en poblaciones de tradición endogámica. Cerca del 60% de los pacientes presentan síntomas antes de las 72 horas de vida, lo que subraya la importancia de un diagnóstico y tratamiento precoces.
Fisiopatología y Base Genética
El HIC se caracteriza por la deficiente regulación de la secreción de insulina en las células β del páncreas, que moviliza la glucosa hacia los tejidos sensibles a la insulina, especialmente al músculo esquelético, tejido adiposo e hígado, provocando hipoglucemia grave. En condiciones normales, cuando los niveles de glucosa disminuyen por debajo de 60 mg/dl, las células β pancreáticas producen pequeñas cantidades de insulina.
La base genética del HIC involucra defectos en genes clave que regulan la secreción de insulina. Hasta la fecha, se han identificado mutaciones en ocho genes diferentes (ABCC8, KCNJ11, GLUD1, CGK, HADH, SLC16A1, HNF4A y UCP2) en pacientes con formas congénitas de hiperinsulinismo. Las formas más graves se deben a mutaciones en los genes ABCC8 y KCNJ11, que codifican los dos componentes de los canales de potasio sensibles al ATP de las células β del páncreas. El daño estructural de estas proteínas o la hiperfunción de enzimas como la glutamato deshidrogenasa (GDH) y la glucocinasa (GK) determinan un estado de despolarización permanente de la célula β, lo que resulta en una secreción insulínica continua que no responde a la concentración de glucemia. Cabe señalar que mutaciones en estos genes se encuentran en aproximadamente el 50% de los casos, sugiriendo la implicación de otros genes aún por identificar.
Desde el punto de vista histológico, el HIC puede presentarse en formas difusa, focal y atípica. La forma difusa es heredada de modo autosómico recesivo o dominante, afectando a todas las células del páncreas. La forma focal es una herencia esporádica, debida a una alteración genética heredada del alelo paterno SUR1 o KIR6.2 asociada a la pérdida espontánea de material genético materno restringida a un foco de células β del páncreas. Los términos "nesidioblastosis" y "nesidiodisplasia", usados anteriormente como sinónimos, han sido declarados como no válidos por muchos autores, ya que tales descripciones se han encontrado también en controles normales.
Manifestaciones Clínicas
El HIC se diagnostica con mayor frecuencia en el período neonatal, presentando signos clínicos que pueden ser graves o inespecíficos. Los signos graves incluyen apneas y convulsiones. Otros signos menos específicos pueden ser rechazo al alimento, irritabilidad, letargo, inestabilidad para termorregular, hipotonía, palidez y apnea. Los recién nacidos con HIC pueden ser macrosómicos debido a la hiperinsulinemia en la vida fetal, especialmente aquellos con mutaciones en el gen HNF4A. Algunos pacientes también pueden presentar miocardiopatía hipertrófica y hepatomegalia.
Diagnóstico del Hiperinsulinismo Congénito
Sospecha y Descarte Diferencial
Los episodios de hipoglucemia deben sospecharse a edades precoces en presencia de signos y síntomas inespecíficos. Durante el período neonatal precoz, es fundamental descartar otras entidades como el hijo de madre diabética, el síndrome de Beckwith-Wiedemann, la asfixia perinatal o la isoinmunización Rh. La recurrencia de los episodios de hipoglucemia debe alertar sobre la posibilidad de un hiperinsulinismo congénito.
Criterios Bioquímicos y Estudios Complementarios
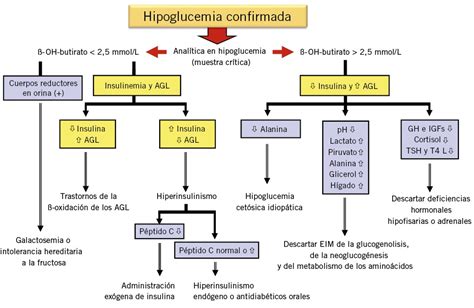
Para confirmar el diagnóstico de HIC, se deben buscar las características bioquímicas que reflejan la acción metabólica de la insulina. Los criterios incluyen: glucemia menor de 40 mg/dl con un índice insulina/glucosa igual o superior a 0,3, ácido β-hidroxibutírico menor de 1 mmol/l y la exclusión de otras causas metabólicas y hormonales. Es frecuente realizar varias determinaciones en momentos diferentes para demostrar la existencia de hiperinsulinismo. En caso de duda diagnóstica, otros parámetros analíticos como el descenso del factor de crecimiento insulinoide (IGF-BP1), una respuesta positiva al glucagón durante la hipoglucemia o una respuesta positiva a la octreótida pueden ser útiles.
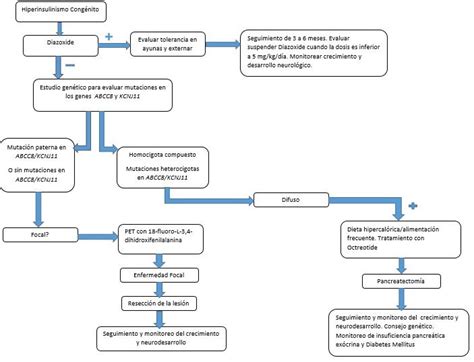
Para confirmar o descartar el HIC, se deben realizar estudios en sangre como la concentración de insulina, glucagón, hormona de crecimiento, cortisol, β-hidroxibutirato y ácidos grasos libres. El diagnóstico puede determinarse basándose en la gravedad de la hipoglucemia que ocurre dentro de las primeras 72 horas de vida. El estudio genético de los padres resulta orientativo, especialmente ante el fracaso del tratamiento inicial con diazóxido.
Síndrome Hiperinsulinismo/Hiperamoniemia (HI/HA)
El síndrome hiperinsulinismo/hiperamoniemia (HI/HA) es una enfermedad genética rara causada por la activación de mutaciones en el gen GLUD1. Se caracteriza por hipoglucemias recurrentes e hiperamoniemia. Un caso reportado describe a un recién nacido pretérmino que presentó llanto débil, hipotonía, dificultad respiratoria leve y episodios recurrentes de hipoglucemia. Los exámenes de laboratorio revelaron amonio sérico elevado (137.6 µmol/L), insulina alta (39.1 µUI/mL), glucemia baja (26.06 mg/dL) y una relación insulina/glucemia de 1.5, lo que permitió el diagnóstico de síndrome HI/HA. Este paciente recibió tratamiento con octreótida y diazóxido, y posteriormente, ante la aparición de crisis convulsivas, se añadieron midazolam, fenobarbital y ácido valproico a su régimen terapéutico.
Manejo Terapéutico del Hiperinsulinismo Congénito
El tratamiento de pacientes con HIC puede ser extremadamente complicado. El objetivo principal es mantener la glucemia en valores mayores a 70 mg/dl para evitar las secuelas neurológicas, ya sea mediante terapia farmacológica o quirúrgica. Es fundamental administrar glucosa por vía intravenosa, junto con alimentación enteral a través de una sonda orogástrica, para asegurar el aporte de carbohidratos. Los pacientes pueden presentar complicaciones como sobrehidratación, insuficiencia cardíaca y sepsis.
Tratamiento Farmacológico
Diazóxido
El diazóxido es el medicamento de primera elección para el tratamiento del HIC. Es un derivado de las benzotiadiazinas, inicialmente desarrollado como antihipertensivo, pero actualmente indicado para la hipoglucemia sintomática por hiperinsulinismo en neonatos. Actúa incrementando la concentración de glucosa sanguínea al inhibir la secreción de insulina por las células β del páncreas, estimulando la liberación de catecolaminas y/o la liberación hepática de glucosa. También hiperpolariza las células del músculo liso arterial activando los canales de K+ sensibles al ATP, lo que induce la relajación del músculo liso vascular.
La dosis debe ser individualizada según la gravedad del cuadro, la glucemia y la respuesta clínica del paciente (dosis inicial de 5 mg/kg/día, máxima de 25 mg/kg/día, dividida en 3-4 dosis por vía oral). Se presenta en cápsulas de 100 mg (Proglicem®), y se recomienda un preparado magistral en suspensión de 10 mg/ml para una mejor dosificación. Como efectos secundarios, el recién nacido frecuentemente presenta retención de agua y sodio (para lo cual se puede asociar hidroclorotiazida en dosis altas), náuseas, vómitos, diarrea, íleo y constipación. Otros efectos incluyen eosinofilia, neutropenia transitoria, trombocitopenia, anemia, taquicardia, cefalea y erupción cutánea. Puede aumentar el metabolismo de la difenilhidantoína.
Octreótida
Si el recién nacido no responde al tratamiento con diazóxido, se recomienda la administración de octreótida, un análogo de la somatostatina. Este medicamento disminuye la traslocación intracelular de iones calcio, lo que inhibe la movilización y liberación de los gránulos de insulina y evita la hipoglucemia. La dosis inicial es de 5 μg/kg cada 6 a 8 horas por vía subcutánea, hasta un máximo de 40 μg/kg/día. Los efectos secundarios más frecuentes incluyen vómitos, diarrea y distensión abdominal, que suelen resolverse espontáneamente dentro de la semana de inicio del tratamiento. No obstante, se ha observado un fenómeno de taquifilaxia que obliga a incrementar la dosis y, en ocasiones, a recurrir a la pancreatectomía.
Otros Fármacos
Los antagonistas de los canales de calcio, como la nifedipina, también han sido empleados a dosis de 0,7-2,5 mg/kg/día.
Tratamiento Quirúrgico: Pancreatectomía
En caso de fracaso del tratamiento médico, se procede a la pancreatectomía subtotal (extirpación del 85-95% del páncreas). Es importante la diferenciación histológica del hiperinsulinismo congénito (forma difusa versus focal) al momento de decidir el tratamiento quirúrgico, ya que esto influye en la extensión de la resección necesaria. Si la hipoglucemia recidiva tras una pancreatectomía subtotal, se realiza un nuevo intento con diazóxido y/u octreótida, y ante una nueva falta de respuesta, se considera la pancreatectomía total (99% de páncreas).
Hiperinsulinismo congénito
Secuelas Neurológicas y su Prevención
Impacto de la Hipoglucemia en el Cerebro Neonatal
La hipoglucemia es una urgencia que debe tratarse rápidamente para minimizar las complicaciones, ya que puede producir daño cerebral permanente con secuelas limitantes para toda la vida e incluso la muerte. El cerebro neonatal es particularmente vulnerable; debido a su tamaño desproporcionadamente grande en relación con la masa corporal, los niños y lactantes tienen una tasa de utilización de glucosa 2 a 3 veces más alta que los adultos. Además, el cerebro tiene solo unos pocos minutos de reservas de combustible en forma de glucógeno, por lo que la interrupción de la entrega de glucosa puede tener consecuencias devastadoras.
La hipoglucemia persistente, especialmente en el contexto de HIC, conlleva un alto riesgo de provocar convulsiones, daño cerebral permanente y retraso en el desarrollo. En estudios clínicos, se ha encontrado que las secuelas neurológicas se presentan en el 28% de los pacientes con HIC, incluyendo retraso psicomotor en grado variable (22%) y epilepsia (13%). Otras alteraciones observadas pueden ser retraso motor fino, déficit de atención, hiperactividad, retraso escolar y retraso del lenguaje. Los primeros meses de vida son el período más vulnerable para el desarrollo de discapacidad, que ocurre en aproximadamente el 25%-50% de los niños con hiperinsulinismo congénito.
Prevención y Evaluación Neurológica
La prioridad es iniciar un tratamiento que mantenga la normoglucemia lo más precoz y eficazmente posible para evitar el daño neurológico. La exposición previa a la hipoglucemia puede atenuar y los episodios repetidos pueden eliminar las respuestas neurogénicas a episodios hipoglucémicos posteriores, lo que lleva a una conciencia reducida o ausente de la hipoglucemia, fenómeno conocido como falla autonómica asociada a hipoglucemia (HAFA).
La evaluación neurológica mediante un análisis de los hitos del desarrollo psicomotor (como el test de Denver) e intelectual (grado de escolarización) es fundamental. La evaluación evolutiva por parte de neurólogos pediatras y psicólogos se lleva a cabo cuando se detecta cualquier anomalía o antecedente de convulsión. Aunque el texto no detalla "escáner cerebral" específico, las técnicas de imagen cerebral son herramientas esenciales para evaluar la extensión del daño neurológico en casos de hipoglucemia persistente y sus secuelas.
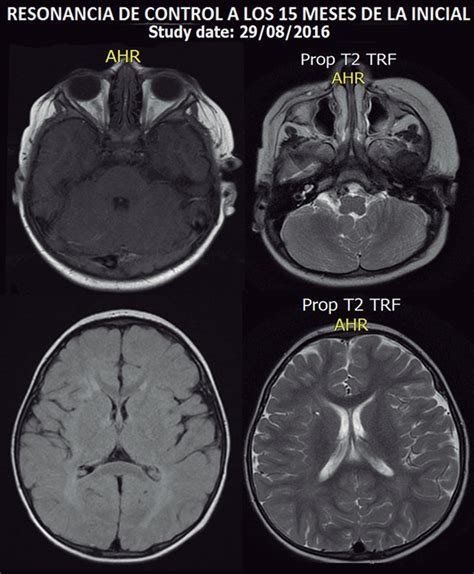
tags: #hipoglucemia #neonatal #escaner #cerebral #hiperinsulinismo