Los medicamentos son creados y sintetizados con el único fin de servir como medidas de curación para patologías y enfermedades. No obstante, muchas veces, durante el proceso de creación de estos, se pasan por alto características inherentes al compuesto que pueden ser perjudiciales para distintos grupos de población.
¿Qué es la talidomida?
La talidomida (N-[2,6-dioxo-3-piperidil]ftalimida) fue sintetizada en 1953 por Wilhem Kunz en los laboratorios Chemie Grünenthal de Alemania. Es un medicamento que cambia la respuesta inmunitaria del cuerpo y reduce su capacidad para desarrollar nuevos vasos sanguíneos. Sus propiedades sedantes e hipnóticas convirtieron tempranamente este fármaco en una buena alternativa a los barbitúricos.
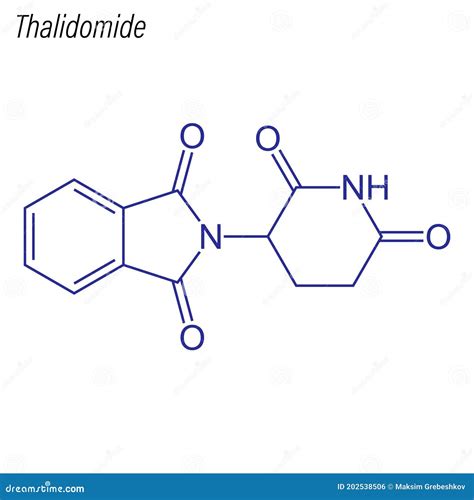
A finales de los años 50, fue comercializada como sustituto o complemento de los barbitúricos, ya que tenía propiedades sedantes e hipnóticas, no producía adicción y carecía de los efectos adversos de los barbitúricos. Gracias a esta característica, su uso comenzó a ser recetado para prevenir las náuseas del embarazo.
Historia de la comercialización
- Cuatro años más tarde, en 1957, se autorizó su venta para el tratamiento sintomático de las náuseas y los vómitos durante el embarazo en Alemania, Canadá e Inglaterra.
- Inmediatamente después, se exportó con más de 80 nombres comerciales a 50 países, con excepción de Francia y Estados Unidos. En estos países, no se autorizó con esta indicación debido a la detección de neuropatía periférica como efecto indeseable.
- En 1959, la talidomida fue comercializada en España formando parte de los principios activos que componían 6 medicamentos: Imidan®, Varilal®, GlutoNaftil®, Softenon®, Noctosediv®, Enero-Sediv® (comprimidos y suspensión). Estos fueron distribuidos por las empresas farmacéuticas Pevya (actualmente UCB Pharma), Medinsa (representante de Chemie Grünenthal) y Farmacobiológicos Nessa.
- El 1 de octubre de 1957, la empresa Chemie Grünenthal GmbH empezó a comercializar la talidomida como un tranquilizante sin receta, bajo el nombre comercial de "Contergan", entre cuyas indicaciones se incluye el uso para las embarazadas contra las náuseas y mareos, y enseguida se convirtió en el tranquilizante más vendido en Alemania.
- En abril de 1958, la empresa Distillers introdujo la talidomida en el mercado del Reino Unido, bajo el nombre de "Distaval", como medicamento sujeto a prescripción médica, cuya indicación principal era precisamente su uso por las mujeres embarazadas para combatir las náuseas y mareos.
- Finalmente, la talidomida se vendió en más de 40 países bajo más de sesenta nombres comerciales distintos.
- En España, la empresa Medina (sucedida por Grünental Pharma) obtuvo autorización administrativa en junio de 1960 y comenzó su venta a partir de noviembre de 1960, bajo el nombre comercial de "Softenon".
Efectos teratogénicos de la talidomida
Lo que no se sabía entonces era que el consumo de dicho medicamento durante el periodo de embarazo podría acarrear grandes consecuencias negativas para el desarrollo del bebé. Es uno de los primeros medicamentos conocidos por causar defectos de nacimiento en humanos.
Malformaciones congénitas asociadas
Poco después de su introducción en el mercado se produjo un incremento espectacular de niños nacidos con focomelia y otras alteraciones genéticas graves. En 1956 se documentó el primer caso aislado de focomelia tras la exposición a talidomida y en los 5 años posteriores se registraron en todo el mundo aproximadamente 3.000 dismelias, malformaciones congénitas extremadamente infrecuentes en los miembros, tales como:
- Amelia: ausencia de todo el miembro.
- Focomelia: pérdida o acortamiento grave de los elementos proximales (con persistencia de los dedos y las manos), por su similitud con la foca.
- Ausencia/hipoplasia del pulgar o los dedos.

Durante esos años, se detectó un aumento progresivo de casos de malformaciones congénitas, con una mortalidad del 40% durante el primer año de vida, que no se limitaban exclusivamente a la afectación de las extremidades, sino que se asociaban también a:
- Alteraciones cardíacas.
- Renales.
- Digestivas.
- Oftálmicas (ojos pequeños o faltantes).
- Auditivas (partes faltantes de los oídos, y la sordera).
- Parálisis de la cara.
- Defectos de los genitales (órganos sexuales).
- Defectos del tracto gastrointestinal (estómago e intestinos).
Además de esto, su uso ha ocasionado miles de muertos, que han podido ser contabilizados. La exposición a talidomida durante el embarazo se ha asociado con un crecimiento deficiente.
Posteriormente, se estableció que el periodo sensible al efecto de la talidomida en el desarrollo embrionario estaba entre los días 20 y 36 después de la fertilización (34-50 días después del último ciclo menstrual). Cuando una persona toma talidomida en esta etapa inicial del embarazo, hay una probabilidad de defectos de nacimiento de al menos el 20%.
Descubrimiento y retirada del mercado
A finales de los años cincuenta y principios de los sesenta, en Alemania, los obstetras y, sobre todo, los pediatras, observaron un aumento dramático de la incidencia de recién nacidos con extremidades acortadas y malformadas, a veces, asociadas a otras alteraciones, especialmente de la región craneal, pero sin sospechar, inicialmente, la causa.
- En 1960 y 1961, Hans Rudolf Wiedemann y Widukind Lenz, pediatras alemanes, y William McBride, un obstetra australiano, de forma paralela y en diferentes países, comenzaron a observar un aumento inusual de casos de malformaciones congénitas severas, particularmente de las extremidades.
- No fue hasta principios de la década de los años sesenta cuando Lenz y McBride descubrieron y denunciaron las anomalías congénitas detectadas en series de recién nacidos cuyas madres habían sido tratadas con talidomida durante el embarazo.
- En 1961, tras la publicación de la carta de Lenz sobre la capacidad teratógena de la talidomida en la revista Lancet, esta fue retirada inicialmente del mercado alemán por Grünenthal y progresivamente en todo el mundo (1961-1962), siendo España uno de los últimos países en prohibirla oficialmente, en enero de 1963.
- La comercialización de medicamentos con talidomida fue prohibida en España mediante la Orden Ministerial de 18 de mayo de 1962. A pesar de que el Colegio de farmacéuticos ordenó a farmacias y mayoristas la retirada de la circulación de productos con talidomida, como el medicamento podía existir en el tráfico o en poder de particulares, se siguieron produciendo afecciones por talidomida hasta el año 1965.
Aunque la cifra de afectados a nivel mundial no se conoce con exactitud, se ha estimado la existencia de más de 10.000 recién nacidos con malformaciones durante el periodo de comercialización de la talidomida. Actualmente, más de 2.000 víctimas de todo el mundo siguen viviendo con las consecuencias de esta tragedia.
Talidomida - Un escándalo farmacéutico | Experimentos Humanos
Mecanismo molecular de la talidomida
Científicos de Alemania han descubierto el mecanismo molecular que provoca los graves defectos de nacimiento que produjo la talidomida en los hijos de mujeres embarazadas que la tomaron. Investigadores de la Universidad Técnica de Munich (TUM) han identificado finalmente el mecanismo molecular de la talidomida, que tiene que ver con la inactivación de un complejo proteínico, que a su vez frena la formación de vasos sanguíneos.
Otros equipos de investigación habían establecido previamente que cereblon, una proteína celular, juega un papel importante en la función de los IMiDs (fármacos inmunomoduladores, como la talidomida, lenalidomida y pomalidomida). Dentro de las células, cereblon se une por lo general a las proteínas CD147 y MCT1. Estas dos proteínas se producen normalmente en la fabricación de sangre y en las células inmunes, y entre otras cosas, promueven la proliferación, el metabolismo y la formación de nuevos vasos sanguíneos. CD147 y MCT1 siempre se producen en pareja, formando un complejo proteínico. Sin embargo, para encontrar su otra mitad y activarse, requieren la ayuda de cereblon. En enfermedades como el mieloma múltiple, un aumento de la cantidad de este complejo proteínico permite a las células tumorales multiplicarse y propagarse rápidamente.
En una célula cancerígena, la talidomida hace que el complejo proteínico (CD147 y MCT1) desaparezca. El resultado es que CD147 y MCT1 ya no se pueden activar y desaparecen. Esta inactivación específica del complejo proteínico provocó los mismos defectos de desarrollo observados después del tratamiento con talidomida. Sin estas dos proteínas, los vasos sanguíneos no pueden desarrollarse adecuadamente. Los resultados de este estudio pueden ayudar a crear nuevas terapias de cáncer sin IMiDs.
Situación actual de los afectados por talidomida en España
Durante más de 30 años, las autoridades españolas negaron la venta de talidomida y, por tanto, la existencia de casos en nuestro país. Aunque se han calculado entre 1.500 y 3.000 los recién nacidos con malformaciones, la falta de un registro oficial de afectados ha impedido el acceso de estos a cualquier compensación económica o ayuda sanitaria. Únicamente 4 españoles fueron recompensados por el gobierno alemán y Grünenthal tras cumplir estrictos criterios médicos, conservar la receta original de la talidomida, el nombre de la farmacia en la que se compró y el propio recipiente.
Reconocimiento y reclamaciones
- En 2003, se fundó la Asociación Española de Víctimas de la Talidomida y otras inhabilidades (AVITE) con el objetivo de reivindicar la situación de este colectivo con la creación de un censo que permitiera un reconocimiento social y económico de los afectados comparable con los de otros países.
- Posteriormente, las autoridades españolas reconocieron la venta legal de talidomida dentro del Sistema Sanitario Público Español entre 1959 y 1965 (3 años después de su prohibición oficial).
- Tras las conversaciones entre la AVITE, el Defensor del Pueblo y el Ministerio de Sanidad y Trabajo, en 2006, el Instituto de Salud Carlos III desarrolló un protocolo clínico para determinar las personas afectadas por la talidomida, a las cuales se les concedió el «certificado de talidomídico» del Centro de Investigación de Anomalías Congénitas.
- El 5 de agosto del 2010, se promulgó el Real Decreto 1006/2010 que reconoce el colectivo afectado por talidomida en España y regula el procedimiento de concesión de ayudas a las personas afectadas. Este contempla como víctimas de la talidomida a los nacidos entre 1960 y 1965, aunque la venta se inició a partir de 1957 y continuó presuntamente hasta 1973, según el vademécum de ese año. Se establecieron ayudas a partir de 30.000 euros para quienes poseían el «certificado de talidomídico» y presentaban una invalidez mínima del 33%. Actualmente, 24 afectados han recibido compensaciones económicas.
- El Real Decreto exigía presentación de la resolución de reconocimiento del grado de discapacidad emitida por el Imserso o por el órgano competente de la respectiva comunidad autónoma, o en su caso, solicitud de reconocimiento del grado de discapacidad, e informe de diagnóstico emitido por el Instituto de Salud Carlos III.
- El importe de la ayuda, entre 30.000 y 100.000 euros, dependía del grado de discapacidad reconocido. Resulta sumamente criticable que el plazo para reclamar una ayuda fuese hasta el 30 de septiembre de 2010, teniendo en cuenta que el BOE se publicó el 6 de agosto de 2010.
Procesos judiciales
- En 2011, los afectados por la talidomida en España, unos 184 reconocidos por la AVITE, interpusieron una demanda colectiva contra Grünenthal y UCB Pharma. Las negociaciones fracasaron cuando Grünenthal ofreció 120.000 euros para compensar a todas las víctimas españolas, es decir, aproximadamente 600 euros por afectado.
- La demanda se presentó finalmente en el Juzgado de primera instancia de Madrid el 12 de febrero de 2012.
- El Juzgado de primera instancia de Madrid, en sentencia de 19 de noviembre de 2013, estimó en parte la demanda, admitiendo como indemnización la solicitada, que es la cantidad resultante de multiplicar por 20.000 euros cada punto porcentual de minusvalía reconocida por la administración española, más los intereses legales.
- Actualmente, la AVITE continúa luchando por conseguir una indemnización de Grünenthal que cubra los gastos médicos, ortopédicos, rehabilitadores y psicológicos derivados de los efectos teratógenos de la talidomida.
En el pasado 9 de septiembre de 2012, poco después del 50.° aniversario de la descripción del efecto teratógeno de la talidomida, Grünenthal, la compañía farmacéutica que la descubrió y comercializó, pidió por primera vez disculpas públicamente por las graves malformaciones provocadas por el fármaco en niños cuyas madres embarazadas tomaron el medicamento. Las palabras utilizadas en el discurso por el director ejecutivo de la compañía han sido consideradas como inapropiadas, insuficientes y tardías por las asociaciones de víctimas de países como Alemania, Gran Bretaña, Japón, Canadá, Australia y España. Todas ellas coinciden unánimemente en que las disculpas carecen de sinceridad, que no se corresponden con la responsabilidad judicial, las irregularidades en el proceso de comercialización de la talidomida y la negativa a proporcionar una compensación económica y recursos sanitarios suficientes a las víctimas.
Nuevas indicaciones de la talidomida y control
Tras la retirada del mercado de talidomida, continuó la posibilidad de administrar el fármaco como sedante e hipnótico exclusivamente para pacientes con lepra en los Estados Unidos.
Usos terapéuticos actuales
En las últimas décadas, el descubrimiento de sus propiedades antiinflamatorias, antiangiogénicas e inmunorreguladoras ha propiciado su utilización como terapia alternativa o de segunda elección en:
- Sarcoidosis.
- Úlcera aftosa en pacientes con virus de la inmunodeficiencia humana (VIH).
- Estomatitis aftosa recurrente.
- Enfermedad de Behçet.
- Prurigo nodular y prurigo actínico.
- Enfermedad de injerto contra huésped crónica.
- Leucemias agudas y síndromes mielodisplásicos.
Finalmente, en 1998, la Food and Drug Administration (FDA) aprobó la comercialización de talidomida (Thalomid®) para el tratamiento del eritema nudoso leproso y posteriormente, en 2006, para el tratamiento del mieloma múltiple en combinación con dexametasona. Ambas aprobaciones fueron sometidas a estrictas recomendaciones y restricciones de dispensación.
En España, la talidomida está incluida desde 1985 entre los medicamentos de especial control médico junto a los derivados de la vitamina A (isotretinoína, acitretina) de administración sistémica, ácido acetohidroxámico, clozapina, pergolida, cabergolina, vigabatrina y sertindol.
Actualmente, está indicada para el tratamiento de primera línea del mieloma múltiple en combinación con melfalán y prednisona, en pacientes con mieloma múltiple no tratado de edad igual o superior a 65 años o no apto para recibir quimioterapia a dosis altas.
Paralelamente, se han sintetizado nuevos inmunomoduladores, derivados estructuralmente de la talidomida, con un perfil similar de teratogenia, como la lenalidomida (Revlimid®, aprobado como medicamento huérfano) y la pomalidomida (CC-4047, en investigación), para el tratamiento de síndromes mielodisplásicos.
Programas de prevención de exposición
Las aprobaciones del uso de talidomida fueron sometidas a estrictas recomendaciones y restricciones de dispensación, destinadas a prevenir y evitar la posibilidad de aparición de malformaciones congénitas, por el programa Sistema para la educación sobre la Talidomida y la Seguridad (Thalidomide REMS®), que incluye:
- Un número limitado de prescriptores y de farmacias.
- Formación y registro de los pacientes.
- Para mujeres con posibilidades de quedar embarazadas, es necesario usar dos métodos anticonceptivos adecuados durante 4 semanas antes de empezar a tomar talidomida, durante el tratamiento y durante 4 semanas después de este.
- Deben tener dos pruebas de embarazo negativas antes de empezar a tomar talidomida y una prueba de embarazo en un laboratorio en ciertos momentos durante el tratamiento.
- Si una mujer queda embarazada durante el tratamiento, el médico debe llamar a la FDA y al fabricante, y hablar con un especialista en problemas del embarazo.
- Para los hombres, la talidomida está presente en el semen. Deberán usar un preservativo sintético o de látex o evitar completamente las relaciones sexuales con una mujer que esté embarazada o pueda quedar embarazada mientras esté tomando este medicamento y durante 4 semanas después del tratamiento.
- No se debe donar sangre ni semen mientras se esté tomando talidomida y por un período de 4 semanas después de terminar el tratamiento.
Implicaciones legales en la regulación y el control de medicamentos
La tragedia de la talidomida obligó a los gobiernos progresivamente a emitir normas y reglamentos que garantizaran la seguridad en el uso de medicamentos, la creación de centros de farmacovigilancia y sistemas para detectar las reacciones adversas de los medicamentos comercializados.
Además, en materia de ensayos clínicos con nuevos medicamentos, supuso el inicio del desarrollo de una estricta normativa sobre productos en fase de desarrollo para garantizar la seguridad y la creación de comités de ética y de investigación para controlar el desarrollo de la investigación clínica en humanos.
Actualmente, la International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH), organismo internacional que reúne a las autoridades sanitarias y las principales industrias farmacéuticas, establece la armonización de los requisitos para la investigación, el registro y la comercialización de nuevos medicamentos. La última guía ICH describe las recomendaciones para la realización de los estudios de toxicidad, carcinogenidad, genotoxicidad y estudios de toxicidad reproductiva y del desarrollo, indispensables para iniciar y proseguir con la fase clínica del desarrollo de un medicamento.
Por otra parte, la experiencia clínica de la talidomida y otros medicamentos en mujeres embarazadas propició en algunos países la elaboración de recomendaciones de uso de los medicamentos durante el periodo de gestación y su clasificación en categorías.
- En 1975, la FDA elaboró la clasificación de medicamentos para su uso en el embarazo en 5 categorías de riesgo (establecido en orden creciente por las letras A, B, C, D o X), de acuerdo con la capacidad de teratogenicidad en función del tipo de estudios realizados y de la información disponible. Esta estratificación del riesgo fetal se ha mantenido hasta el 2010, con la elaboración de una nueva clasificación integral de los medicamentos durante la gestación y lactancia (Pregnancy and Lactation Labeling), que aún no ha entrado en vigor.
- Existen 2 sistemas más de clasificación internacional del riesgo fetal, el Swedish Catalogue of Approved Drugs (categorías A, B1, B2, B3, C, D) y el australiano, Australian categorisation system for prescribing medicines in pregnancy (A, B1, B2, B3, C, D, X), que combina los 2 sistemas previos y cuyas categorías no corresponden exactamente con las de la FDA.
- En España, se dispone de la Guía de prescripción terapéutica, adaptación del British Nacional Formulary, que incluye un apéndice específico sobre el embarazo y los fármacos que deben evitarse o usarse con precaución durante este período.
Actualmente, los resultados de la investigación preclínica y clínica, de acuerdo con las pautas para la evaluación de medicamentos de la EMA, se resumen en la ficha técnica autorizada del medicamento. En España, los profesionales sanitarios y las mujeres embarazadas pueden realizar consultas sobre teratógenos al Servicio de Información Telefónica sobre Teratógenos Español y al Servicio de Información Telefónica para la Embarazada, respectivamente.
¿Podría volver a producirse una tragedia similar a la de la talidomida?
La devastadora experiencia con la talidomida fue el punto de partida en la percepción de los riesgos de los medicamentos y, en consecuencia, de la implantación de un marco de protección científico, ético y legal. Ahora, 50 años después de esta tragedia, tras el desarrollo de estrictas regulaciones en la investigación preclínica y clínica, recomendaciones sobre el uso de fármacos en embarazadas y los sistemas de vigilancia de defectos congénitos e identificación de teratógenos, resulta difícil imaginar una nueva epidemia como la que produjo la talidomida.
De todas maneras, factores como el aumento de la edad de concepción, la mayor prevalencia de enfermedad crónica en el embarazo o la posibilidad de diagnóstico y tratamiento de la afección fetal, podrían favorecer la prescripción de determinados fármacos, algunos de ellos con mecanismos de acción poco conocidos y, consecuentemente, podría aumentar el riesgo potencial de teratogenia. Sin embargo, la historia (p. ej., el caso del dietilestilbestrol y el aumento del riesgo de adenocarcinoma de células claras en fetos mujeres) ha demostrado que todavía queda un largo camino para aprender qué tratamientos son los más seguros y eficaces durante el embarazo.
tags: #efectos #secundarios #de #la #talidomida #en