La Hemoglobina: Una Molécula Fundamental
La hemoglobina (Hb) es una proteína globular crucial, presente en los hematíes en altas concentraciones. Su función vital es la participación en el intercambio gaseoso, fijando oxígeno en los pulmones para transportarlo por la sangre hacia los tejidos y células que rodean el lecho capilar del sistema vascular. Al volver a los pulmones, desde la red de capilares, la hemoglobina también actúa como transportador de CO2 y de protones.
La gran variedad de aspectos científicos que incluye la hemoglobina y la importancia que juega en la biología hacen que, aunque los primeros estudios se realizaron desde el siglo XIX, aún hoy aparezcan sorprendentes descubrimientos acerca de esta molécula, tales como las nuevas globinas (neuroglobina y citoglobina) y sus llamativas interacciones con el óxido nítrico. Asimismo, el estudio de las hemoglobinopatías constituye un gran reto para la medicina moderna, en la medida en que ponga al servicio de sus pacientes los resultados de la investigación científica básica.
Importancia Histórica y Científica
La hemoglobina ha jugado un papel histórico en la química, la biología y la medicina. En 1849, se convirtió en la primera proteína en ser cristalizada y asociada con una función fisiológica específica. La diferencia morfológica entre los cristales de hemoglobina de diferentes organismos proporcionó por primera vez evidencia contundente acerca de la especificidad en la expresión proteica entre las especies. Además, se encuentra entre las primeras proteínas cuyo peso molecular fue determinado correctamente. En 1958, se convirtió en la primera proteína eucariota en ser sintetizada in vitro, trabajo que permitió comprobar que el mecanismo de síntesis proteica en eucariotas es similar al de Escherichia coli. Su estructura se estableció en 1960. El ARN mensajero de la globina fue el primer mensajero eucariota en ser aislado y en tener una secuencia nucleótida determinada.
El descubrimiento de que la anemia de células falciformes es causada por el reemplazo de uno solo de los 287 residuos de aminoácidos, presentó por primera vez indicios de que una mutación puntual en un gen estructural puede causar la sustitución de un aminoácido en la proteína codificada por este gen y, consecuentemente, enfermedad. De igual forma, la transición de la síntesis de hemoglobina desde la vida fetal a la adulta es un gran ejemplo de diferenciación celular.
Genética y Biosíntesis de la Hemoglobina
La biosíntesis de la Hb guarda estrecha relación con la eritropoyesis. La expresión genética y el contenido de Hb acompañan la diferenciación de las unidades formadoras de colonias eritroides (UFC-E) en precursores eritroides.
Genes de la Globina y su Localización Cromosómica
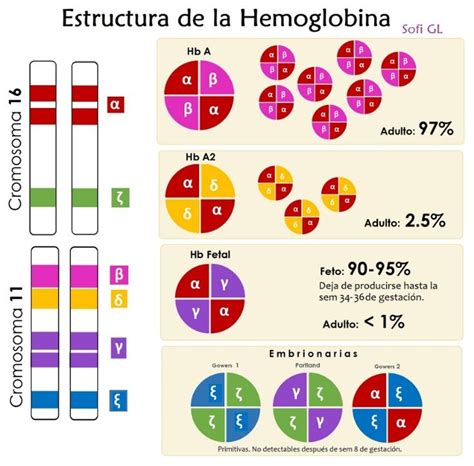
Cada una de las cadenas polipeptídicas de la Hb cuenta con genes propios: alfa (α), beta (β), delta (δ), gamma (γ), y épsilon (ε). Los genes α y β son independientes y se ubican en cromosomas distintos. El grupo de genes α se localiza en el brazo corto del cromosoma 16 y contiene además los codificadores de la cadena zeta (ζ). El grupo de genes β se localiza en el brazo corto del cromosoma 11 e incluye a los genes de las cadenas Gγ, Aγ, δ y ε.
Todos los genes funcionales de la globina comparten una estructura general que consiste en 3 exones (secuencias codificadoras) y 2 intrones o sectores interpuestos (secuencias que no se traducen). La región promotora incluye alrededor de 100 pares de bases que preceden al punto de comienzo de la transcripción (punto de clivaje). Tres secuencias de esta región se fijan a la ARN polimerasa que cataliza la síntesis de ARN mensajero. Existen dos secuencias claves en la iniciación de la transcripción: TATA y CAT; las mutaciones que las afectan limitan la transcripción de ARNm. La porción distal del tercer exón (AATAAA) finaliza la transcripción. Solamente entre 5% a 10% del material genético de los eritroblastos se transcribe; los genes de la globina pertenecen a esta fracción.
Proceso de Transcripción y Traducción
La síntesis de ARN se lleva a cabo bajo la influencia de grupos enzimáticos denominados ARN polimerasas. La transcripción primaria del ARNm incluye copias de toda la secuencia del ADN genómico (intrones y exones). Antes de su transporte al citoplasma, se procesa por clivaje del extremo 5', hay separación de las secuencias transcriptas de los intrones y poliadenilación del extremo 3'. Este último paso es esencial en el transporte y estabilización citoplasmática del ARNm. La separación implica la formación de asas en el pre-ARNm, de manera que los extremos distales de los exones (puntos dadores) se acerquen a los proximales de los subsiguientes exones (puntos receptores). Luego, los intrones sufren clivaje enzimático y los puntos dadores y receptores se sellan. Los puntos de consenso son secuencias de nucleótidos adyacentes que perfeccionan la síntesis del ARNm. Las mutaciones que involucran tanto los puntos de unión, así como los de consenso, alteran la separación y crean ARNm anormales.
La causa más común de las hemoglobinopatías es la mutación puntual, es decir, la sustitución de un nucleótido de ADN por otro, lo que modifica el código genético y puede inducir un cambio en un aminoácido de la globina resultante. Por ejemplo, en la anemia de células falciformes (HbS), el ácido glutámico se reemplaza por una valina en el aminoácido 6 de la cadena β: β6 (A3) Glu ----> Val.
La traducción es un proceso ribosómico en donde se sintetiza una cadena polipeptídica de acuerdo con un patrón proporcionado por la secuencia de codones del ARNm. Incluye cuatro etapas:
- Activación: Se forma el ARN de transferencia (ARNt).
- Iniciación: El ARNt que contiene metionina se alinea con el codón iniciador AUG en el ARNm del ribosoma.
- Elongación: Cada anticodón del ARNt se adosa a cada codón del ARNm. Los aminoácidos del ARNt se adosan mediante un puente peptídico a otro aminoácido ya unido al ribosoma.
- Terminación: Se produce cuando se llega a un codón de finalización UAA, la cadena polipeptídica se completa y se separa del ribosoma.
Los polipéptidos libres forman de inmediato dímeros αβ y tetrámeros α2β2.
Regulación de la Eritropoyesis y Estabilización de Cadenas
La maduración de proeritroblastos a eritrocitos está controlada positivamente por la hormona polipeptídica eritropoyetina, que promueve tanto la proliferación como la sobrevida de los precursores eritroides. También, sobre la superficie de dichos precursores, se encuentran unos receptores que promueven la apoptosis y que son estimulados por enzimas denominadas caspasas. La activación de estos receptores (o la deprivación de eritropoyetina) conduce al clivaje, por parte de las caspasas, de una proteína regulatoria nuclear llamada GATA-1, indispensable para el proceso eritropoyético, lo que conduce a apoptosis y detención de la maduración. Los receptores de "muerte celular" se han denominado sistema Fas/FasL.
La constante investigación acerca de los genes sobre los cuales actúa el factor de transcripción genético GATA-1 condujo al descubrimiento de un gen expresado en altos niveles en el eritroblasto, que codifica una proteína chaperona denominada AHSP (proteína estabilizante de cadena alfa), la cual se une específicamente a las cadenas α. Su papel está relacionado con la estabilización de dichas cadenas, evitando que se precipiten formando inclusiones citoplasmáticas (cuerpos de Heinz) que afectan a la membrana celular y conducen a la lisis eritrocitaria.
Síntesis del Grupo Hemo
El grupo hemo se sintetiza en virtualmente todos los tejidos, pero su síntesis es más pronunciada en la médula ósea y el hígado, debido a la necesidad de incorporarlo en la Hb y los citocromos, respectivamente. Es una molécula plana que consta de un hierro ferroso y un anillo tetrapirrólico, la protoporfirina IX. Característicamente, demuestra una banda a 440 nm o de Soret y otras cuatro más en el espectro visible. El hemo es un factor fundamental en la regulación de la tasa de síntesis de la globina. Su principal efecto se ejerce en la iniciación de la traducción, donde bloquea la acción de un inhibidor de la producción de globina. También participa en la transcripción y el procesamiento del ARNm. Su papel en la síntesis proteica en los mamíferos se extiende más allá del eritrocito; en el tejido hepático y cerebral se demuestran sustancias que dependen del hemo para comenzar la producción de proteínas.
Normalmente, los eritrocitos envejecidos se degradan hacia el día 120 de vida en la médula ósea, el hígado y el bazo. En algunas circunstancias, sin embargo, los eritrocitos sufren lisis intravascular, liberando Hb, que puede ser tóxica para los tejidos a menos que se remueva rápidamente. La haptoglobina (Hp) es una proteína plasmática que une Hb libre, a través de la formación de un complejo Hp-Hb. Este complejo es reconocido a través de una proteína situada en la superficie de los macrófagos y monocitos denominada CD163, permitiendo su digestión y la seguida liberación de hierro y bilirrubina. La expresión de Hp y CD163 está regulada por proteínas de fase aguda como la interleucina 6 (IL-6), sugiriendo que las enfermedades inflamatorias crónicas se relacionan con alteraciones del metabolismo del hierro.
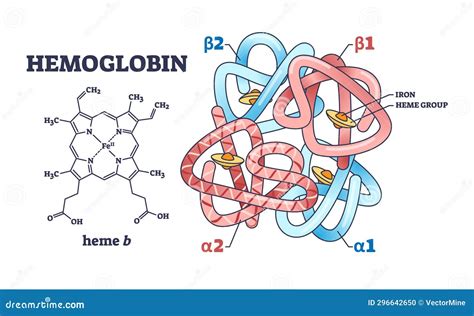
Estructura de la Hemoglobina
La hemoglobina es una proteína con estructura cuaternaria, lo que significa que está constituida por cuatro cadenas polipeptídicas, cada una con un grupo prostético hemo.
Composición del Grupo Hemo
Un grupo prostético es la porción no polipeptídica de una proteína en su estado funcional. En el caso de la hemoglobina, el grupo prostético es el hemo, un tetrapirrol cíclico que les proporciona el color rojo a los hematíes. El hemo es una molécula de porfirina, específicamente la protoporfirina IX, que contiene dos grupos ácidos propiónicos, dos vinilos y cuatro metilos como cadenas laterales unidas a los anillos pirrólicos de la estructura de la porfirina.
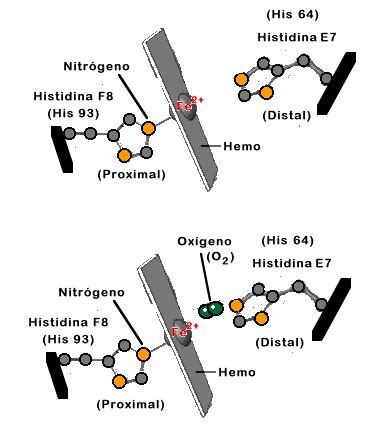
El átomo de hierro se encuentra en estado de oxidación ferroso (+2) y puede formar cinco o seis enlaces de coordinación dependiendo de la unión del O2 (u otro ligando) a la Hb (oxiHb, desoxiHb). Cuatro de estos enlaces se producen con los nitrógenos pirrólicos de la porfirina en un plano horizontal. El quinto enlace de coordinación se realiza con el nitrógeno del imidazol de una histidina denominada histidina proximal. Finalmente, el sexto enlace del átomo ferroso es con el O2, que además está unido a un segundo imidazol de una histidina denominada histidina distal. Tanto el quinto como el sexto enlace se encuentran en un plano perpendicular al plano del anillo de porfirina.
Cadenas Polipeptídicas y Estructura Cuaternaria
Las cuatro cadenas polipeptídicas de la Hb contienen cada una un grupo hemo. Las cadenas polipeptídicas α contienen 141 aminoácidos, mientras que las no α (β, γ, δ) contienen 146 aminoácidos y difieren en la secuencia de aminoácidos. Se conoce desde hace décadas la estructura primaria de las cuatro cadenas de Hb normales.
La estructura secundaria es muy similar: cada una exhibe 8 segmentos helicoidales designados con las letras A a la H. Entre ellos se encuentran 7 segmentos no helicoidales: NA, AB, CD, EF, FG, GH y HC. Esta distinción es fundamental, pues los segmentos helicoides son rígidos y lineales, mientras que los no helicoidales son flexibles. Como el hierro del hemo forma un puente covalente con la histidina proximal (F8) y el O2 se une de forma covalente al hemo y a la histidina distal (E7), el hemo queda suspendido en una hendidura no polar entre los helicoides E y F.
La parte porfirínica del hemo se sitúa dentro de una bolsa hidrofóbica que se forma en cada una de las cadenas polipeptídicas. Las estructuras obtenidas por difracción de rayos X muestran que en la bolsa del hemo existen unas 80 interacciones entre 18 aminoácidos y el hemo. La mayoría de estas interacciones no covalentes se presentan entre cadenas apolares de aminoácidos y las regiones no polares de la porfirina.
Aunque cada cadena α está en contacto con las cadenas β, existen pocas interacciones entre las dos cadenas α o entre las dos cadenas β entre sí. Los principales contactos intercatenarios son α1β1 y α1β2, que determinan dos estructuras cuaternarias: una para la oxiHb y otra para la desoxiHb. Cuando una proteína está con su grupo prostético se denomina holoproteína, y cuando está sin este, se la denomina apoproteína. Además, por poseer un grupo prostético, la Hb es una proteína conjugada, una hemoproteína.
Transición de la Síntesis de Hemoglobina Durante el Desarrollo
Uno de los ejemplos más llamativos de la relevancia del proceso evolutivo y la eficiencia de los sistemas biológicos se encuentra en la transición de la síntesis de hemoglobina desde la vida embrionaria y fetal hasta la adulta. Este patrón de síntesis es esencial para adaptarse a las diferentes condiciones de oxigenación a lo largo del desarrollo.
Hemoglobinas Embrionarias
En el feto humano, en un principio, no se sintetizan cadenas α ni β, sino las cadenas zeta (ζ) y épsilon (ε). Fisiológicamente, se forman tres variantes transitorias embrionarias:
- Hb Gower 1: Compuesta por dos cadenas ζ y dos cadenas ε (ζ2ε2).
- Hb Gower 2: Compuesta por dos cadenas α y dos cadenas ε (α2ε2).
- Hb Portland: Compuesta por dos cadenas ζ y dos cadenas γ (ζ2γ2).
Estas hemoglobinas embrionarias predominan en las primeras semanas de gestación. Al final del primer trimestre, las subunidades α han reemplazado a las subunidades ζ, y las subunidades γ han reemplazado a los péptidos ε, marcando el inicio de la predominancia de la hemoglobina fetal.
Hemoglobina Fetal (HbF)
La hemoglobina fetal (HbF), con una composición de dos cadenas α y dos cadenas γ (α2γ2), representa el 90-95% de toda la hemoglobina a las 34-36 semanas de gestación. A término, la HbF representa el 53-95% de toda la hemoglobina, debido a su mayor afinidad por el oxígeno, crucial para la extracción de oxígeno de la sangre materna. El porcentaje de hemoglobina F permanece estático durante las 2 primeras semanas de vida y luego disminuye aproximadamente un 3% por semana cuando se reinicia la eritropoyesis. Normalmente, es inferior al 2-3% de la hemoglobina total a los 6 meses de edad. Además de aumentar en algunas hemoglobinopatías, los niveles de HbF pueden incrementarse en bebés pequeños para la edad gestacional, que han sufrido hipoxia crónica o que tienen trisomía 13.
Hemoglobina del Adulto (HbA y HbA2)
La hemoglobina del adulto, HbA, (α2β2) representa el 4-13% de la hemoglobina total del feto. Después de las 34 semanas de gestación, la producción de HbA aumenta significativamente a medida que disminuye la de HbF. La HbA se convierte en la hemoglobina predominante a los 3 meses de edad, aunque este cambio puede retrasarse en los recién nacidos prematuros enfermos.
La hemoglobina A2 (α2δ2) se produce en pequeñas cantidades desde el nacimiento y suele alcanzar los niveles adultos a los 6 meses de edad, aunque puede seguir aumentando durante los 1-2 primeros años de vida. La HbA2 y la Hb Bart's pueden detectarse en bebés normales nacidos a término.
El patrón de síntesis de la hemoglobina durante el desarrollo explica por qué las anomalías de la cadena α causan problemas clínicos desde las primeras etapas de la vida fetal, mientras que las anomalías de la cadena β pueden ser difíciles de diagnosticar en el periodo neonatal debido a la predominancia inicial de HbF.
Sintesis de la hemoglobina (fisiologia del globulo rojo) Formacion de la Hb.
Variantes de Hemoglobina y Hemoglobinopatías
Fisiológicamente, se forman seis variantes de Hb humana:
- Tres variantes transitorias embrionarias: Hb Gower 1, Hb Gower 2 y Hb Portland.
- HbF, que es la que predomina en la vida fetal.
- HbA, que constituye más del 95% de la Hb del adulto.
- HbA2, también presente en un porcentaje menor (1-3,5%) en el niño y en el adulto.
Las cadenas de globina que forman la molécula de Hb posnatalmente se asocian en forma de un tetrámero y se designan como α, β, γ y δ. Así, las variantes de Hb se forman por las siguientes combinaciones de cadenas:
- HbA: α2β2
- HbF: α2γ2
- HbA2: α2δ2
Hay muchas variantes de Hb determinadas genéticamente (heredadas) y, aunque la mayoría son inocuas, otras ocasionan alteraciones clínicas notables.
Categorías de Hemoglobinopatías
Las hemoglobinopatías pueden agruparse en tres categorías fundamentales:
- Variantes estructurales de la Hb: Como la HbS (anemia falciforme o drepanocitosis). Se atribuyen a la sustitución de aminoácidos en las cadenas de globina. Se han descrito casi un millar, pero la mayoría son un hallazgo casual y clínicamente silentes. Las que ocasionan enfermedad se deben a la sustitución de aminoácidos que conlleva cambios en la estructura secundaria o terciaria del tetrámero de Hb.
- Fallo en la síntesis de una cantidad adecuada de Hb normal (talasemias): Por defecto en la producción de una o más de las cadenas de globina. La clínica asociada surge de la combinación de una inadecuada síntesis de Hb y de la acumulación de cadenas de globina que no han podido formar el tetrámero. Lo primero causa anemia y microcitosis, y lo segundo, eritropoyesis ineficaz y hemólisis.
- Fallo en el salto normal que se produce en el neonato de producir HbF a HbA (persistencia hereditaria de Hb fetal).
Para el diagnóstico de estas alteraciones, la interpretación de diversas pruebas de laboratorio es crucial, de las cuales la más relevante es la electroforesis (EF) de Hb. La electroforesis de hemoglobina a pH alcalino es un método simple, rápido y sensible que detecta las variantes de Hb más comunes, separando las que tienen distintas cargas en su superficie. Sin embargo, puede tener dificultades para detectar bandas menores o diferenciar ciertas hemoglobinas en neonatos. La electroforesis a pH ácido es muy efectiva en combinación con la anterior. Actualmente, la cromatografía líquida de alta resolución (HPLC) es muy útil para cuantificar de forma rápida HbA2 y HbF, así como para identificar variantes estructurales, siendo un método imprescindible para analizar gran cantidad de muestras en poco tiempo, como en el cribado neonatal de anemia falciforme.
tags: #sintesis #de #hemoglobina #embrionaria