Introducción al Piebaldismo
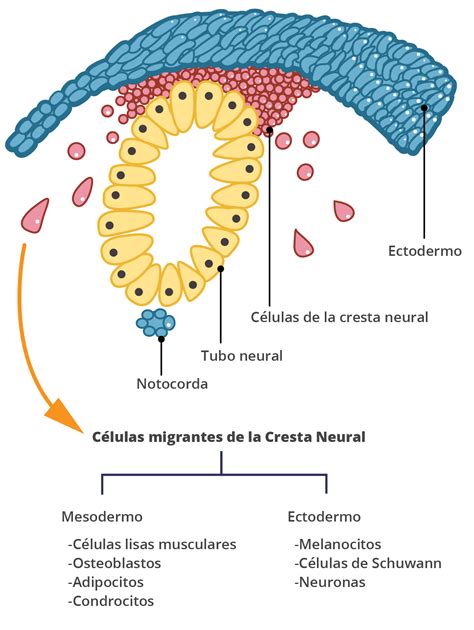
La pigmentación de la piel es un factor esencial determinado por la deposición de **melanina** en las células epidérmicas. Las células dendríticas que producen el pigmento melánico, conocidas como **melanocitos** o **melanoblastos**, provienen de la cresta neural del embrión y migran durante el periodo fetal para colonizar la piel, los pelos, los ojos, la mucosa, las leptomeninges y el oído interno. El color de la piel está genéticamente controlado por la melanina epidérmica, junto con otros pigmentos como la hemoglobina oxigenada, la hemoglobina desoxigenada y los depósitos de carotenos o bilis no metabolizada.
Las alteraciones en la pigmentación se relacionan con la disminución (leucodermia) o el aumento (melanodermia) de la misma. Se denomina **hipopigmentación** o **hipomelanosis** a una disminución localizada o generalizada del color de la piel, generalmente causada por una reducción de la melanina en la epidermis. Las hipomelanosis verdaderas pueden ser de tipo melanopénico (número normal de melanocitos con melanina reducida, como en el albinismo) o melanocitopénico, debido a la disminución o ausencia de melanocitos, como ocurre en el **piebaldismo**.
El piebaldismo es una **leucodermia congénita**, estable a lo largo del tiempo y de herencia **autosómica dominante**. Es un trastorno infrecuente, con una incidencia que no supera 1:20.000 a 1:100.000 personas, afectando por igual a hombres y mujeres, sin predominio de razas.
Bases Genéticas del Piebaldismo
El defecto genético principal del piebaldismo se ha identificado en el gen **KIT**, ubicado en el cromosoma 4q12. Este gen codifica para un receptor transmembrana de la tirosina quinasa, una proteína esencial para la proliferación, diferenciación y migración de los melanoblastos (precursores de los melanocitos) desde la cresta neural hacia la epidermis durante la embriogénesis.
La proteína KIT también es importante para las células germinales y las sanguíneas. Su ligando es un factor de crecimiento embrionario, también conocido como *mast cell growth factor* o *stem cell growth factor*. La activación del receptor por su ligando estimula la migración y proliferación de los melanoblastos, un evento indispensable para el desarrollo normal de los melanocitos.
Las mutaciones en el gen KIT pueden llevar a una expresión reducida del receptor transmembrana y, por lo tanto, a una disminución de la proliferación y una distribución anómala de los melanoblastos, lo que resulta en la aparición de manchas sin pigmento. Se han reportado 14 mutaciones puntuales, 9 deleciones, 2 mutaciones en desplazamientos de nucleótidos y 3 inserciones relacionadas con el piebaldismo. La **expresión fenotípica** de la enfermedad es muy variable y se correlaciona con el tipo y localización de la mutación dentro del gen que codifica la proteína c-Kit. Por ejemplo, mutaciones específicas pueden resultar en fenotipos severos, mientras que otras en el dominio extracelular pueden generar fenotipos moderados a leves.
Manifestaciones Clínicas en Recién Nacidos
Clínicamente, el piebaldismo se caracteriza por un fenotipo distintivo de parches congénitos de **piel blanca (leucodermia)**, acompañados por un **mechón de pelo blanco frontal (poliosis)**, que puede extenderse a la parte media de las cejas y las pestañas. Estas manchas o mechones amelanóticos (sin color) son, en la mayoría de los casos, los únicos hallazgos visibles de la enfermedad.
- El **mechón de pelo blanco** está presente en el 80% al 90% de los pacientes, es congénito, a menudo triangular, elongado o en forma de diamante, y se ubica en la línea media de la región frontal.
- Las **máculas hipocrómicas** o despigmentadas suelen ser de distribución simétrica, localizadas típicamente en la frente, el tronco (especialmente la porción anterior y central), y las extremidades (desde la mitad de los brazos hasta las muñecas y desde la mitad de los muslos hasta la mitad de las piernas).
- Ocasionalmente, pueden observarse máculas hiperpigmentadas pequeñas dentro de las áreas acrómicas, que no están uniformemente pigmentadas y tienen una apariencia característica.
A diferencia del vitíligo, la hipopigmentación en el piebaldismo es **congénita y estática** en tamaño y forma. Sin embargo, con el tiempo o la exposición solar, pueden aparecer pequeños islotes de hiperpigmentación dentro de las máculas hipopigmentadas.
En la mayoría de los casos, los pacientes con piebaldismo son por lo demás sanos y no presentan manifestaciones extracutáneas, aunque la condición puede generar alteraciones psicosociales.
La piel del recién nacido aspectos clínicos esenciales para su cuidado
Diagnóstico y Diagnóstico Diferencial
El diagnóstico del piebaldismo es fundamentalmente **clínico**, basándose en la presencia de las características máculas de leucodermia y el mechón blanco frontal desde el nacimiento. El estudio histopatológico de las zonas despigmentadas revela una ausencia completa de melanocitos y melanina.
Dada la importancia de descartar otras condiciones, el enfoque diagnóstico debe ser **multidisciplinario**, incluyendo evaluación oftalmológica, neurológica, otorrinolaringológica y genética.
El **diagnóstico diferencial** es crucial para distinguir el piebaldismo de otras hipomelanosis, incluyendo:
- Síndrome de Waardenburg (SW1): Hereditario, se caracteriza por poliosis, sordera neurosensorial congénita, heterocromía del iris, sinofris, raíz nasal alta y amplia, y desplazamiento lateral del canto interno.
- Albinismo: Engloba un grupo de enfermedades genéticas que causan una pérdida parcial o completa del color de la piel, los ojos o el pelo, a menudo con estrabismo, nistagmo o ceguera. Se distinguen el albinismo oculocutáneo (OCA1, OCA2) y el síndrome de Hermansky-Pudlak (albinismo con trastorno hemorrágico y posibles patologías pulmonares e intestinales).
- Vitíligo: Generalmente adquirido, tiene su incidencia máxima en la tercera década de la vida, es inestable y no heredado (aunque puede recurrir familiarmente).
- Síndrome de Woolf: Trastorno autosómico recesivo similar al piebaldismo en sus características pigmentarias cutáneas, pero se diferencia por la presencia de sordera.
- Hipomelanosis de Ito: Un trastorno que causa manchas hipopigmentadas que siguen las líneas de Blaschko.
- Síndromes de Chediak-Higashi y Griscelli: Entidades autosómicas recesivas con inmunodeficiencia grave y alteraciones neurológicas.
Manejo y Tratamiento
Actualmente, no existe un tratamiento médico específico para curar el piebaldismo. El manejo se centra en:
- Fotoprotección: Es fundamental proteger las áreas acrómicas de la exposición solar para evitar quemaduras y el riesgo de cáncer de piel, ya que carecen de melanina.
- Opciones cosméticas: Se pueden emplear camuflaje cosmético, maquillaje o tintes capilares para disimular las áreas despigmentadas, especialmente el mechón blanco. La dihidroxiacetona (DHA) también se puede utilizar como agente pigmentador de la piel.
- Tratamientos quirúrgicos: En casos seleccionados, se han explorado técnicas como los injertos o el trasplante de melanocitos autólogos cultivados *in vitro*, con resultados variables. La fototerapia por sí sola tiene poco efecto, pero puede ser útil después de trasplantes. El metoxaleno oral y tópico puede inducir nuevas manchas hiperpigmentadas dentro de las lesiones, aunque los resultados cosméticos no siempre son satisfactorios.
- Consejo genético: Debido al patrón de herencia autosómico dominante, el consejo genético es esencial para las familias afectadas, informando sobre el riesgo de transmisión a la descendencia (50% de probabilidad en cada embarazo).
Consideraciones Especiales: Piebaldismo y Síndromes Asociados
Aunque el piebaldismo clásico se considera una condición benigna y sin otras complicaciones de salud, algunas mutaciones genéticas o la coincidencia con otros trastornos pueden generar alteraciones adicionales. Se ha descrito que, en ciertos casos, esta condición puede generar otro tipo de alteraciones en el cuerpo, como esterilidad, anemia o defectos en el desarrollo de las neuronas, el intestino o el oído. Sin embargo, estas no son características típicas del piebaldismo en sí, sino de síndromes más complejos donde mutaciones en genes relacionados con el gen KIT o su vía de señalización están involucradas.
Un ejemplo de asociación poco frecuente, pero que ha sido reportado en recién nacidos, es la coexistencia de **piebaldismo con Síndrome de Moebius**. El síndrome de Moebius es una parálisis congénita del nervio facial (VII par craneal), que puede estar asociada a compromiso de otros pares craneales (frecuentemente el VI o abducens) o de otros sistemas, como defectos en las extremidades. La aparición de estas anomalías sugiere una alteración de la morfogénesis entre las semanas 4 y 7 de gestación.
En algunos casos complejos donde se presenta piebaldismo junto con el Síndrome de Moebius, se ha explorado una posible asociación con la exposición prenatal a **misoprostol**. Este fármaco, un análogo sintético de la prostaglandina E1, se ha relacionado estadísticamente con un aumento en el riesgo de ocurrencia de la secuencia de Moebius, artrogriposis y defectos de tipo terminal y transversal de las extremidades cuando se utiliza durante el primer trimestre del embarazo. Sin embargo, la relación de causalidad entre el misoprostol y las malformaciones congénitas sigue siendo controversial y no bien definida, especialmente en el contexto del piebaldismo, que es de etiología genética.
tags: #piebaldismo #recien #nacido