La osteogénesis imperfecta (OI), también conocida como la enfermedad de los huesos de cristal, es un conjunto heterogéneo de trastornos de origen genético que afectan la integridad del tejido conectivo, impidiendo que el cuerpo fabrique unos huesos fuertes y, consecuentemente, provocando que se deformen y fracturen con facilidad, en ocasiones, sin motivo aparente. Esta patología presenta distintos niveles de gravedad, lo que se traduce en diversos grados de afectación a lo largo de la vida del paciente.
Causas de la Osteogénesis Imperfecta
La osteogénesis imperfecta ocurre debido a una mutación, es decir, un cambio genético, en los genes que codifican las cadenas pro-alfa del procolágeno tipo I. El colágeno tipo I es una proteína presente en todos los tejidos de sostén, especialmente en el hueso (donde es el principal componente de la matriz orgánica), piel, tendones, ligamentos, fascias, córnea, esclera, dentina y vasos sanguíneos.
Las mutaciones se presentan en la síntesis del colágeno, ya sean autosómicas dominantes o recesivas. Los niños pueden heredar la mutación de uno de sus padres; sin embargo, en ocasiones, esta mutación no es hereditaria y ninguno de los padres presenta la afección. Los niños con osteogénesis imperfecta nacen con esta condición, lo que significa que no tienen suficiente colágeno en los huesos o su colágeno no funciona como debería, resultando en huesos más débiles y frágiles de lo normal.
Se han descrito más de 200 mutaciones diferentes de los genes que codifican para el colágeno tipo I, afectando aproximadamente en el 90% de los casos al gen que codifica la cadena α1 (COL1A1 en el cromosoma 17) y la cadena α2 (COL1A2 en el cromosoma 7). Estas mutaciones se dividen en dos grandes grupos:
- Las asociadas a interrupción prematura del proceso de traslación, que conlleva haploinsuficiencia por deleción funcional o inactivación del alelo mutado, produciendo un colágeno estructuralmente normal, pero en cantidad reducida. Esto ocurre en la osteogénesis imperfecta tipo I, la forma más leve.
- Otras mutaciones originan defectos estructurales en una de las cadenas α. En el 85% de los casos son mutaciones puntuales que causan la sustitución de glicina por otro aminoácido, distorsionando la estructura normal de triple hélice. Estas mutaciones tienen un efecto dominante negativo sobre el alelo no mutado, con lo que se sintetiza una proteína de colágeno aberrante, en menor cantidad y susceptible de mayor degradación. Esto es lo que ocurre en las formas graves de osteogénesis imperfecta (tipos II, III y IV).
Aunque la OI es un trastorno genético de herencia autosómica dominante, la existencia de "mosaicismo parental" explica que el riesgo de recurrencia para una pareja sana de tener otro hijo afectado pueda ser de hasta el 5-8%.
Clasificación y Manifestaciones Clínicas
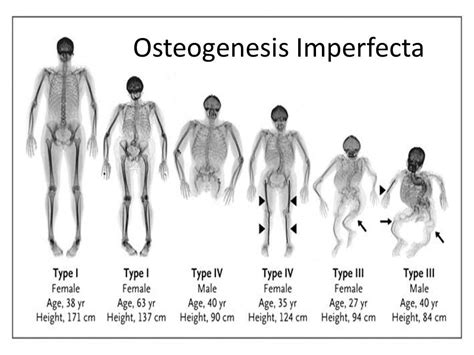
Los síntomas de esta enfermedad varían de una persona a otra. La osteogénesis imperfecta es muy heterogénea en cuanto a presentación, con manifestaciones clínicas muy diferentes, lo que implica que algunas personas están muy afectadas y otras tienen una afectación moderada o leve. En función de la clínica y las alteraciones radiológicas, Sillence (1979) estableció una clasificación en cinco tipos que aún permanece en vigor:
Tipo I: Leve (Escleróticas Azules)
- Es la forma más frecuente y la más leve.
- Los pacientes tienen un colágeno normal, pero en menor cantidad de lo habitual.
- Los huesos son frágiles y se rompen con más facilidad, aunque no suelen presentar deformidades óseas.
- La primera fractura suele ocurrir cuando el niño empieza a andar. Las fracturas disminuyen típicamente después de la pubertad.
- El blanco de los ojos (esclerótica) puede tener un tinte azulado.
- Aparece sordera neurosensorial en un 50% de los casos.
- Se divide en subtipos: IA (sin alteración de la dentinogénesis) y IB (con alteración de la dentinogénesis).
- Talla final normal o con ligero retraso del crecimiento.
- Modo de herencia autosómica dominante (HAD), incidencia de 1/30.000 nacidos vivos.
Tipo II: Letal Perinatal (Neonatal Letal)
- Es la forma más grave de osteogénesis imperfecta.
- El colágeno no se forma con normalidad y la fragilidad ósea es extrema.
- Los bebés con este tipo de OI suelen nacer con muchas fracturas, son muy pequeños y tienen graves problemas respiratorios.
- Radiológicamente se observan costillas arrosariadas, huesos largos "arrugados", osteopenia difusa e islotes óseos en la bóveda craneal.
- Las escleróticas son azules.
- El cráneo es blando y, al palparlo, produce la sensación de una bolsa de huesos. El traumatismo durante el parto puede provocar hemorragia intracraneal y muerte fetal, o los recién nacidos pueden presentar muerte súbita durante los primeros días o semanas de vida por insuficiencia pulmonar.
- Suele ocurrir la muerte perinatal por insuficiencia pulmonar.
- Herencia HAD, mutaciones de novo o por mosaicismo de los padres. Incidencia de 1/20.000-1/60.000 nacidos vivos.
Caso Clínico con Evidencia Neonatal
Se ha reportado un caso de un paciente masculino de un día de vida extrauterina, producto de una madre de 20 años, primigesta y prima segunda de su pareja. Nació por cesárea debido a presentación pélvica, con líquido amniótico meconizado y un peso al nacer de 2275 gramos (RNTPEG). Al examen físico, el niño se encontraba flácido, con cianosis leve, fontanelas amplias con comunicación de la anterior con la posterior, ausencia de escama occipital, escleras azules, retrognatia, extremidades cortas y con crepitación al movimiento. Las radiografías óseas revelaron fracturas múltiples, formación de callo óseo y cambios displásicos en metáfisis. Los genetistas diagnosticaron osteogénesis imperfecta (OI) y se ofreció consejo genético a los padres. Este caso ilustra claramente la presentación grave y la evidencia de OI en el período neonatal.
Tipo III: Deformante Progresiva
- Es la forma no letal más grave de osteogénesis imperfecta, con un colágeno que no se forma con normalidad.
- Los bebés suelen nacer con fracturas.
- Cuando crecen, los huesos se rompen con facilidad y tienden a tener una estatura significativamente menor que sus compañeros.
- Pueden presentar malformaciones o anomalías óseas importantes (cifoescoliosis, deformidades torácicas y de extremidades), dolor crónico e incapacidad funcional.
- La talla final es muy baja.
- Las escleróticas pueden ser azules al nacer y luego se normalizan.
- Generalmente existe alteración de la dentinogénesis y la sordera es rara.
- Herencia HAD, mutaciones de novo, mosaicismo en los padres o herencia autosómica recesiva (rara).
Tipo IV: Moderada
- Es de gravedad intermedia.
- El colágeno no se forma con normalidad.
- Los niños se rompen los huesos a menudo (pudiéndose reducir estas fracturas a partir de la pubertad) y pueden tener huesos de formas anormales (similar a la OI de tipo III).
- La tasa de supervivencia es alta.
- Por lo general, las escleróticas son de color normal (azules al nacer y luego mejoran).
- La talla es moderada-baja.
- Presenta subtipos: IA (sin alteración de la dentinogénesis) y IB (con alteración de la dentinogénesis).
- La sordera es rara.
- Herencia HAD o mutaciones de novo frecuentes.
Tipo V: Moderada o Intensa
- Descrita recientemente.
- Existe limitación de la prono-supinación del antebrazo, laxitud ligamentosa y formación de callos hiperplásicos en las fracturas.
- No se alteran las escleróticas ni la dentinogénesis.
- Herencia HAD.
Otras consecuencias de la mutación del colágeno tipo I a otros niveles son: hiperlaxitud de ligamentos y tendones, fragilidad vascular, disminución de la fuerza muscular, y alteración de la dentinogénesis.
El 50-65% de los pacientes con osteogénesis imperfecta presenta hipoacusia, que puede aparecer en cualquiera de los tipos principales.
Complicaciones Secundarias
Las deformidades de los huesos y los defectos del colágeno comunes a la enfermedad pueden afectar varios órganos internos, lo que provoca problemas secundarios, entre los que se encuentran las complicaciones pulmonares, cardíacas o neurológicas, que pueden desembocar en procesos muy graves por las infecciones, la propia formación de los tejidos afectados o la mal función valvular cardíaca.
Existe una complicación grave denominada impresión basilar, provocada por la malformación de la columna vertebral que presiona la médula espinal y el tallo cerebral. Si no se trata, su progresión puede provocar un rápido deterioro neurológico e impedir respirar.
Diagnóstico de la Osteogénesis Imperfecta
El diagnóstico de la OI es fundamentalmente clínico y radiológico, pero no hay ningún criterio estandarizado. La atención temprana de la patología es importante con el fin de minimizar los signos y síntomas y mejorar la calidad de vida de los pacientes. Se sigue utilizando la clasificación según Sillence, en base a la clínica, hallazgos radiológicos y tipo de herencia.
El diagnóstico exacto es crucial, ya que estos pacientes pueden beneficiarse del tratamiento. Los métodos diagnósticos incluyen:
- Evaluación clínica: Historia de fracturas, deformidades óseas, cifoescoliosis, retraso del crecimiento, escleras azules, dentinogénesis imperfecta, hipoacusia, y antecedentes familiares.
- Hallazgos radiológicos: Presencia de fracturas, osteopenia (disminución de densidad y contenido mineral óseo) variable, rarefacción ósea progresiva, callos múltiples de distinta antigüedad. Los pacientes pueden desarrollar cambios quísticos, densos o frágiles.
- Ecografía de nivel II: Permite la detección intrauterina de osteogénesis imperfecta grave.
- Análisis del procolágeno tipo I o pruebas genéticas: Cuando el diagnóstico es dudoso, puede recurrirse al análisis del procolágeno tipo I de fibroblastos cultivados (obtenidos por biopsia de piel) o al análisis de la secuencia de los genes de COL1A1 y COL1A2. La biopsia de piel confirma el diagnóstico al demostrar un patrón electroforético del colágeno tipo I diferente del normal, aunque en un 10% de los casos la mutación se presenta solo a nivel de hueso y la biopsia puede ser normal.
- Marcadores bioquímicos del metabolismo óseo: Existe disminución de los marcadores de formación y resorción ósea, pero en las formas graves predomina la resorción.
- Densitometría ósea (DEXA): Valora el contenido mineral óseo a nivel L1-L4. En las formas graves la densidad mineral ósea (BDM) es baja o muy baja, pero algunas formas muy leves de OI pueden tener BMD normal.
- Biopsia ósea: Las alteraciones morfológicas y ultraestructurales guardan estrecha correlación con la gravedad clínica, proporcionando información que no se puede obtener de otra manera y ayudando a clasificar de una forma más exacta los distintos grupos de OI.
Opciones Terapéuticas
En la actualidad, no existe ningún tratamiento curativo para la OI, ya que no se puede actuar directamente sobre la formación de colágeno tipo I. El tratamiento es sintomático y debe enfocarse de manera multidisciplinaria, dirigido a la prevención y control de los síntomas, la mejora de la movilidad independiente y el desarrollo de una masa ósea y fuerza muscular óptimas. El pronóstico vital depende de la gravedad de la presentación y del buen manejo que se haga de la enfermedad.
Tratamiento Médico
- Hormona de crecimiento (GH): Actúa estimulando la proliferación celular (osteoblastos) y la síntesis de matriz extracelular, facilitando la aposición y mineralización ósea. También aumenta la fuerza y la masa muscular, y favorece la recuperación de las fracturas. La GH estaría indicada en OI con defecto cuantitativo en la síntesis de colágeno tipo I estructuralmente normal (formas leves o tipo I), pero no en OI con alteración estructural del colágeno I (formas más graves, sobre todo la tipo III) porque se produciría mayor cantidad de colágeno anómalo.
- Bifosfonatos: Son fármacos que se acumulan en el hueso y se adsorben a la superficie de los cristales de hidroxiapatita, reduciendo la solubilidad de la matriz ósea y haciéndola más resistente a la acción de los osteoclastos. Se usa pamidronato intravenoso o alendronato oral para aumentar la densidad ósea y disminuir el dolor y el riesgo de fractura. Se ha observado que el tratamiento con pamidronato muestra una mejor respuesta cuanto más precozmente se utiliza, sobre todo en niños menores de 2 años.
- Denosumab: Es un potente inhibidor de la resorción ósea osteoclástica y suele administrarse por vía inyectable. Los estudios han mostrado que este medicamento es beneficioso en algunos pacientes con osteogénesis imperfecta.
- Suplementos: Se debe administrar un aporte adecuado de calcio oral y de vitamina D a las personas con deficiencia de esta hormona.
Tratamiento Combinado de GH y Pamidronato
Este tratamiento favorece la ganancia de contenido mineral óseo por mecanismos distintos y complementarios, ya que la GH favorece fundamentalmente la formación ósea y el pamidronato inhibe la resorción ósea. Un estudio realizado en el Servicio de Endocrinología Infantil del Hospital La Paz con 11 pacientes con OI graves (7 casos tipo III y 4 casos tipo IV) mostró una gran mejoría clínica con la combinación de GH y pamidronato. Se observó una disminución o ausencia del número de fracturas, mejor consolidación de las existentes, estabilización de las deformidades, disminución o ausencia de dolores óseos, y una mejora importante de la calidad de vida. Aunque no se observó mejoría de la talla ni de la velocidad de crecimiento en la mayoría de los casos, sí se aumentó la densidad mineral ósea (BMD) de manera significativa. Como efectos secundarios del pamidronato se encontraron fiebre o febrícula, malestar general, náuseas/vómitos, dolores articulares y un caso autolimitado con hipertensión arterial, apareciendo estas complicaciones en el primer ciclo de tratamiento.
OrtoPedCast 18 - Principios de Osteogénesis Imperfecta - Entrevista a Oswaldo Lazala
Fisioterapia y Terapia Ocupacional
- Fisioterapia: Ayuda a desarrollar la fuerza muscular, aumentar las aptitudes físicas generales, mejorar la respiración y aprender a usar ayudas y dispositivos para desplazarse, si son necesarios.
- Terapia ocupacional: Ayuda a los niños a usar dispositivos asistenciales, si son necesarios.
- Los cuidados de las fracturas y el soporte ortopédico son esenciales. El uso de sillas de ruedas, de apoyos y de otras ayudas para la deambulación es frecuente en estos pacientes.
Cirugía
La cirugía se utiliza para corregir los huesos rotos o malformados. En algunos casos de hipoacusia, está indicado el implante coclear.
Rol de los Padres
Con su apoyo, el niño puede recibir los cuidados médicos necesarios para llevar un estilo de vida lo más saludable posible. Es fundamental ayudar al hijo a encontrar actividades y aficiones que sean seguras para él y con las que disfrute, revisando con el médico los deportes que debe evitar. Mantenerse activos como familia, escogiendo actividades que el hijo pueda hacer, contribuye a su bienestar. Tener un hijo con una enfermedad de por vida puede ser estresante, por lo que el apoyo parental es clave.
tags: #osteogenesis #imperfecta #evidencia #en #el #periodo